Animal experiments
All male mice were on a C3H background and purchased from Beijing Vital River Laboratory Animal Technology. All mice were bred in a specific-pathogen-free (SPF) environment with free access to food and water and housed in standard conditions of 20–26 °C temperature and 40–70% humidity under a 12 h light cycle at Guangzhou Medical University. All animal experiments protocols used in this study were approved by the Animal Care Committee of the Guangzhou Medical University (GY2022144). Consistent with previous research [7], the Control mice (n = 3) were fed with a complete standard Control diet. For MDBs model mice, the DDC-Fed mice (n = 3) were fed the Control diet and added 0.1% DDC (Sigma-Aldrich, 137030-25G) for 10 weeks to induce the formation of MDBs. They were then withdrawn from DDC for 1 month (n = 3), at which time the MDBs had mostly disappeared. They were then refed DDC (n = 3) for 6 to 10 days as was previously done [7]. Then the liver tissue from each group was obtained for subsequent experiments: (1) liver tissue was subjected to nuclei isolation and snRNA-seq; (2) Tissue was cut into distinct small sections and preserved in 4% polyformaldehyde for subsequent immunostaining; (3) liver tissue and serum were preserved at -80 °C for RNA and protein lysates extracting, ELISA, qPCR and Western blot analysis.
Patients and clinical specimens
Human formalin-fixed paraffin-embedded (FFPE) liver biopsies from patients with HCC (n = 3) were obtained from the Qingyuan Affiliated Hospital of Guangzhou Medical University archives, with Institutional Review Board approval (IRB-2022-094). The patients exhibited features of MDBs on liver histopathology and the details were shown in Table 1. The liver biopsy sections were 4 μm thick. The study was carried out according to the principles of the Declaration of Helsinki and the data were analyzed anonymously and reported.
Table 1 Liver and serum parameters in 3 HCC samples which had formed MDBsSingle-nucleus RNA-Sequencing (snRNA-seq) and bioinformatics
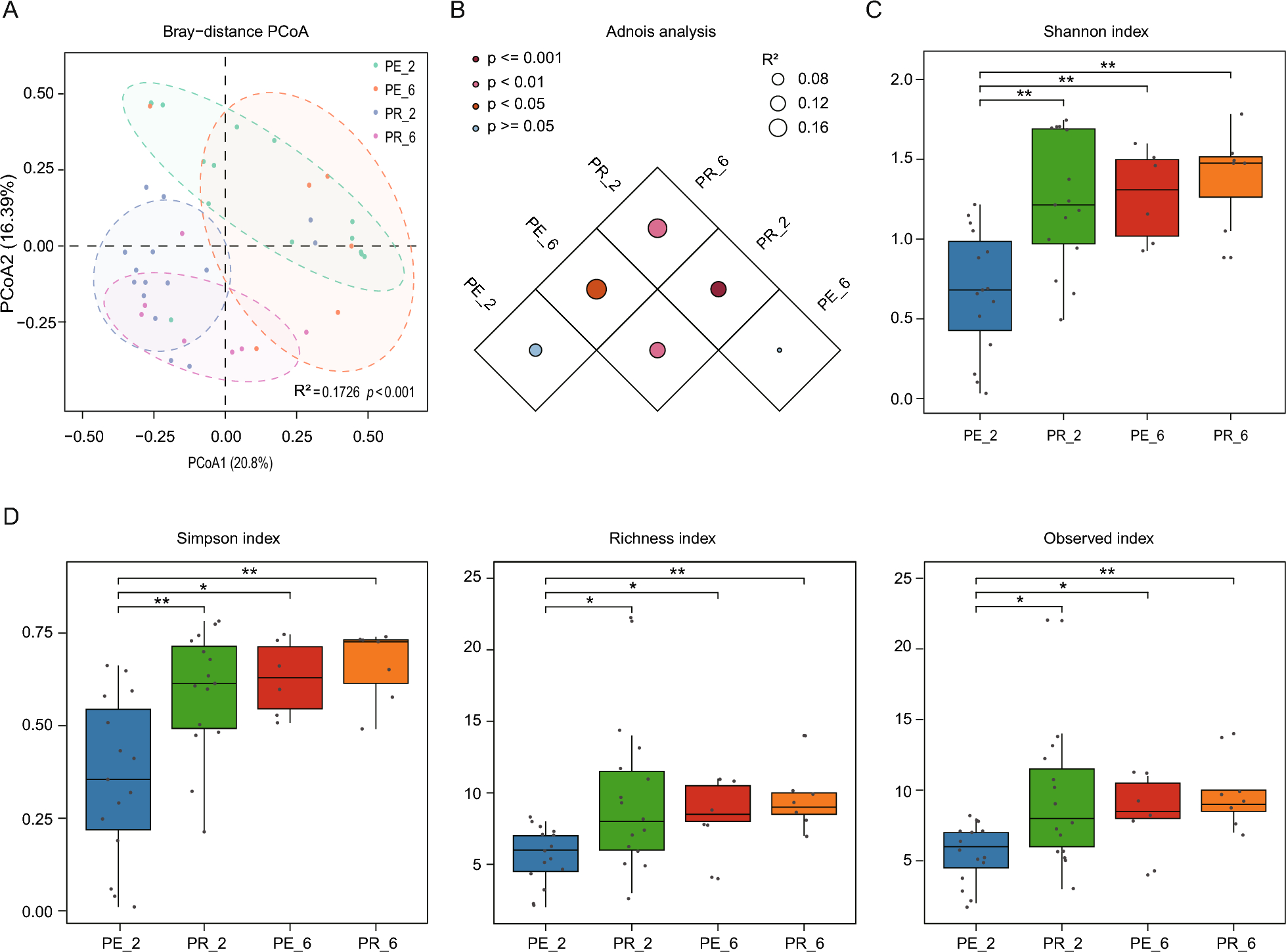
Nuclei isolation and snRNA-seq dataset of 10× Genomics® as standard protocol as described previously [28] and data analysis was mainly performed by Genedenovo Biotechnology Co., Ltd (Guangzhou, China). Clustering analysis composed of R package Seurat was used to perform quality control and exploration of snRNA-seq data. Then, Seurat continued to use Uniform manifold approximation and projection (UMAP) to visualize and explore these datasets. The differentially expressed gene analysis and expression value of each gene in the given cluster were compared against the rest of cells using the Wilcoxon rank sum test. Kyoto Encyclopedia of Genes and Genomes (KEGG) was used to further understand the biological functions of genes and pathway enrichment analysis. Gene set variation analysis (GSVA) was performed by the “GSVA” R package for GSVA enrichment analysis.
Hematoxylin and eosin (H&E) staining
H&E staining was performed following a standard protocol for mice liver tissue sections. Briefly, liver sections were deparaffinized with TO and then hydrated in decreasing concentrations of alcohol for subsequent staining. The liver sections were stained in hematoxylin solution for 3 min and then washed in PBS of 0.1% Tween-20 for 5 min. Next, the slides were put into eosin solution for 2 min to stain the cytoplasm. After washing for 5 min in tap water, the sections were dehydrated and then a drop of mounting medium was added to mount each of the liver tissue. Images were collected by slide scanner systems PRECICE 500 (UNIC) and processed by iViewer software (UNIC).
Immunohistochemical assay
Liver sections were deparaffinized with TO and hydrated in decreasing concentrations of alcohol. Antigen epitope retrieval was performed by microwave heating within a citrate antigen retrieval solution (Beyotime, P0081). The sections were incubated with 0.3%H2O2 for 15 min and blocked in PBS of 5%BSA for 1 h at room temperature (RT). Next, sections were stained with mouse anti-F4/80 (Bioss, bsm-34028 M) or rabbit anti-IL-7R (Bioss, bs-1540R) overnight at 4 °C. After washing, sections were incubated with HRP-conjugated secondary antibodies for 1 h. Then, the signal was detected using a DAB kit (ZSGB-Bio, ZLI-9018). At last, the sections were counterstained with hematoxylin. Images were collected by slide scanner systems PRECICE 500 (UNIC) and then processed by iViewer software (UNIC).
RNA extraction and quantitative PCR (qPCR)
Total cells and tissue RNA were extracted as indicated using Cell/Tissue Total RNA Kit (Yeason, 19221ES50) and then reverse-transcribed into cDNA using 5×HiScript III qRT SuperMix (Vazyme, R323-01). qPCR was performed using TB Green® Premix Ex Taq™ II (TaKaRa, RR820A) with the primers listed in Table 2.
Table 2 Sets of specific oligonucleotide quantitative real-time PCR (qRT-PCR) primersImmunofluorescence
After deparaffinization and antigen retrieval in darkness, liver sections were blocked in PBS of 5% BSA for 1 h at RT. Autofluorescence was quenched using a tissue autofluo quencher (Servicebio, G1221), followed by washing PBS of 0.1% Tween-20 three times (3 min/per). Sections were incubated with rabbit anti-F4/80; rabbit anti-IL-7R, rabbit anti-GPNMB (Bioss, bs-2684R); rabbit anti-8-OHdG (Bioss, bs-1278R); rabbit anti-ASC (CST, #67824); rabbit anti-KRT8 (Abcam, ab55407 (Red); or proteintech, 27105-1-AP (Green)), mouse anti-nucleoporin p62 (Santacruz, sc-48373) at 4 °C overnight. Sections were washed and incubated with Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen, A11008), Alexa Fluor 488 goat anti-mouse IgG (Invitrogen, A11001), Alexa Fluor 555 goat anti-rabbit IgG (Invitrogen, A21428), Alexa Fluor 555 goat anti-mouse IgG (Invitrogen, A21422) and Alexa Fluor 647 donkey anti-rabbit IgG (Invitrogen, A31573) for 1 h. The sections were then stained with DAPI (Beyotime, c1002) for 5 min and washed in PBS of 0.1%Tween-20 for 5 min three times before mounting with Antifade Mounting Medium (Beyotime, P0128M). Images were observed and snapped by laser confocal microscope (ZEISS, LSM880) equipped with photomultiplier tube (PMT) detectors. The pictures were then processed by ZEN software (ZEISS) at a resolution of 1024 × 1024 pixels.
Transmission electron microscopical analysis
For transmission electron microscopy, the liver tissue from the Control and DDC-Fed mice were sliced into 1 mm3 sections fixed with 2.5% glutaraldehyde (PH7.4) for 2 h. After washing with 0.1 M phosphate buffer (pH 7.2) for 3 times, the tissue were then fixed in 1% osmic acid for 2 h at 4℃. Next, the sections were hydrated in decreasing concentrations of alcohol and subsequently embedded in Epon-Araldite resin (Ted Pella Inc). The ultrathin sections were cut using a Leica EM UC7 Ultramicrotome (Leica, LeicaUC7) and then counterstained with 3% uranyl acetate and 2.7% lead citrate. The sections were observed using a JEM1400 transmission electron microscope (JEOL, JEM1400) and operated at 80 kV.
MDBs induction in vitro
This experiment was repeated and improved the long-term treatment of hepa1-6 described previously [14]. 8 × 106 cells were seeded in 12-well Cell Culture Plates or 10-cm cell culture dishes. The hepa1-6 cells were continuously co-stimulated with 40ng/ml TNFα (PeproTech, 315–01 A) and 400ng/ml IFNγ (novoprotein, C746) for 10 days with intervals of three days. The cytokine and medium were refreshed every three days, then the cells were harvested for immunofluorescence and qPCR at day 10.
Raw264.7/Hepa1-6 contact co-culture and mitochondrial labelling
Raw264.7 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, 11965092) containing 1% penicillin/streptomycin and 10% FBS (Excell, FSS500). Hepa1-6 were cultured in DMEM (NAHCO3 concentration of 1.5/L, iCell-128-000) supplemented with 5% FBS. All cells were cultured under a humidified atmosphere with 5% CO2 at 37 °C. Raw264.7 cells were seeded in 12-well chambered cell culture slides (CITOTEST) for 12 h before initiation of the co-culture. Hepa1-6 cells and MDB-forming hepa1-6 cells were seeded in 6-well plates at a density of 150,000 cells per well in a 1:1 ratio and stained for 30 min using Mitotracker (Invitrogen, M7512) after culturing overnight for draining the excess dye. Then the stained hepa1-6 cells were added to the preseeded Raw264.7 cells and contact co-cultures were maintained for 24 h in DMEM with 1.5 g/L NAHCO3.
Western blot
Proteins extracted from the liver tissues by RIPA lysis buffer (Beyotime, P0013B) with protease cocktail inhibitor Ι (Apexbio, K1007). Lysates were mixed with 5× SDS Loading buffer and boiled at 100 °C for 10 min. Then, they were separated on SDS-PAGE (10%polyacrylamide) and transferred to polyvinylidene fluoride membranes (PVDF) (Millipore, ISEQ00010). The proteins solution were mixed with anti-NLRP3 rabbit mAb (Abcam, EPR23094-1), ASC rabbit mAb, Pro-Caspase-1 rabbit mAb (MCE, HY-P80587), Cleaved-Caspase-1 rabbit mAb (MCE, HY-P80622), IL-1β rabbit mAb (Abclonal, A16288), β-actin mouse mAb (Proteintech, HRP-66009), α-tubulin (Beyotime, AT-819-1) and GAPDH mouse mAb (Proteintech, 60004-1-Ig). HRP-conjugated secondary antibodies were used for protein detection, followed by exposure to the chemiluminescence imaging system Pxi9 (Syngene) and then images were processed with ImageJ software.
Enzyme-linked immunosorbent assay (ELISA)
This analysis was performed following a standard protocol provided by ELISA kit (Thermo, 88–7013 A-88). The optical density (OD) of all samples to be tested (liver protein lysates and cell culture supernatant) was determined by VARIOSKAN FlASH (Thermo) at 450 nm. The data were processed by GraphPad Prism version 9.0 (GraphPad Software, California, USA).
Raw264.7/Hepa1-6 Transwell coculture system
Total 1 × 106 Control hepa1-6 or MDBs hepa1-6 cell model described above was added to the upper chamber of Transwell-24 inserts (6.5 mm diameter with polycarbonate membrane filters containing 3 μm pores, Nest, CN) and allowed them to attach for 12 h at 37 °C and 5% CO2. The medium was removed and replaced with 100 µL of DMEM (NaHCO3 concentration of 1.5/L with 5% FBS) to support cell survival, and then the transwell inserts containing hepa1-6 cells were transferred to Raw264.7 macrophage-seeded wells (n = 3 per phenotype). LPS-primed Raw264.7 cells were stimulated by 100ng/ml LPS (Sigma, L2630) for 24 h to initiate macrophage activation. The lower compartment contained high glucose DMEM supplemented with 10% FBS. The cells and supernatants were harvested after coculturing for 24 h and were lysed with 350 µl LB Buffer (Yeasen, CN) and subjected to qPCR and ELISA analysis.
Statistical analysis
The data of all quantitative in vitro and in vivo experiments (three mice per group) are represented as mean ± SEM or SD of three independent experiments. P-value < 0.05 was considered statistically significant. All statistical analyses using the GraphPad Prism version 9.0 (GraphPad Software, California, USA).
Data Availability
SnRNA-seq data generated in this work have been deposited in the GEO with assigned accession numbers GSE201569. These data will be also available from the corresponding author upon request.







留言 (0)