
記住我






The human hepatocellular carcinoma cell line, HepaRG (Biopredic International, Saint Grégoire, France), was maintained in an undifferentiated state using HepaRG cells culture medium for 2 weeks (proliferation phase), which consisted of William’s Medium E (AL240, Omni Life Science) supplemented with 10% fetal calf serum (FCS; 10270-106, Life Technologies), 5 µg/mL insulin (Novo Nordisk), 2 mM glutamine (M11-006, Sigma), 50 µM hydrocortisone (Pfizer), and 1% Penicillin/Streptomycin (Pen/Strep; P0781, Sigma). The cells were incubated in a humidified cell incubator at 37 °C and 5% CO2, with medium changes performed three times a week. Differentiation of the cells was initiated by the addition of 1.7% dimethyl sulfoxide (DMSO; 4720.2, Roth).
SCP-1, a mesenchymal stem cell line (kindly provided by Prof. Dr. Matthias Schieker (Bocker et al. 2008)) was used as osteoprogenitor cells and cultured in Minimum Essential Medium Eagle Alpha (MEM α; AL081A, Omni Life Science) supplemented with 5% FCS at 37 °C in a humidified atmosphere with 5% CO2. The growing medium was refreshed twice weekly.
The human leukaemia monocytic cell line, THP-1 cells (ACC16, DSMZ) were used as the osteoclastic precursor cells, which were cultured as a suspension cell culture in RPMI 1640 Medium (R8758, Sigma) supplemented with 5% FCS. The cells were maintained in a humidified atmosphere with 5% CO2 at 37 °C, and the growing medium was renewed twice weekly.
Human platelet-rich plasma (hPRP) scaffolds manufacturing and sterilisationPRP scaffolds were generated as previously described (Haussling et al. 2019). To prepare the PRP, EDTA blood samples were collected from a group of five healthy volunteers and then centrifuged at 1000 × g for 10 min. A mixture was then prepared by combining an aqueous solution containing 16.0% pHEMA (128635-500G, Sigma), 0.3% Bis-Acrylamide (3039.1, Carl Roth), and 0.25 g/L PRP, this mixture was carefully stirred and cooled in ice. Following 30 min of incubation, di-sodium hydrogen phosphate buffer (T876.1, Carl Roth) was added to the solution to achieve a final concentration of 0.3 M. Immediately after this step, a mixture of 0.1% glutaraldehyde (3778.1, Carl Roth), 0.2% Ammonium Persulfate (A3678-25G, Sigma), and 0.2% TEMED (2367.3, Carl Roth) was added and thoroughly mixed. The resulting solution was then dispensed into polystyrene casting molds, with each mold receiving 2 mL of the solution, and then immediately frozen at a temperature of -18 °C for at least 12 h. To facilitate the slicing of the formed matrix with a razor blade, it was deep-frozen at a temperature of -80 °C for 1 h. The resulting scaffolds had uniform dimensions, measuring 3 mm in height and 6 mm in diameter. Afterward, the scaffolds were immediately transferred to a 1 M CaCl2 (CN93.2, Carl Roth) solution to promote the crystallisation of calcium phosphate, specifically hydroxyapatite. Following a 24-h incubation period, the CaCl2 solution was carefully removed, and the scaffolds were washed with phosphate-buffered saline (PBS; L182-50, Merck) for 15 min. During this process, ice crystals were formed to serve as placeholders for creating the desired pores. The size and shape of the pores were affected by the temperature and speed at which the freezing occurs (Adnan Memic and Joseph Steingold 2019).
To eliminate the unreacted compounds that may potentially be toxic and to also achieve sterilisation, the PBS was completely removed from the scaffolds. Subsequently, the scaffolds were immersed in 70% ethanol and shaken for at least 12 h. Following this, the sterilised scaffolds underwent four washing steps using sterile PBS for 30, 60, 90, and 120 min, respectively. Afterward, the scaffolds were incubated in THP-1 cell growing medium under humidified conditions with 5% CO2 at a temperature of 37 °C for 48 h. This pre-conditioning process served as a sterility control.
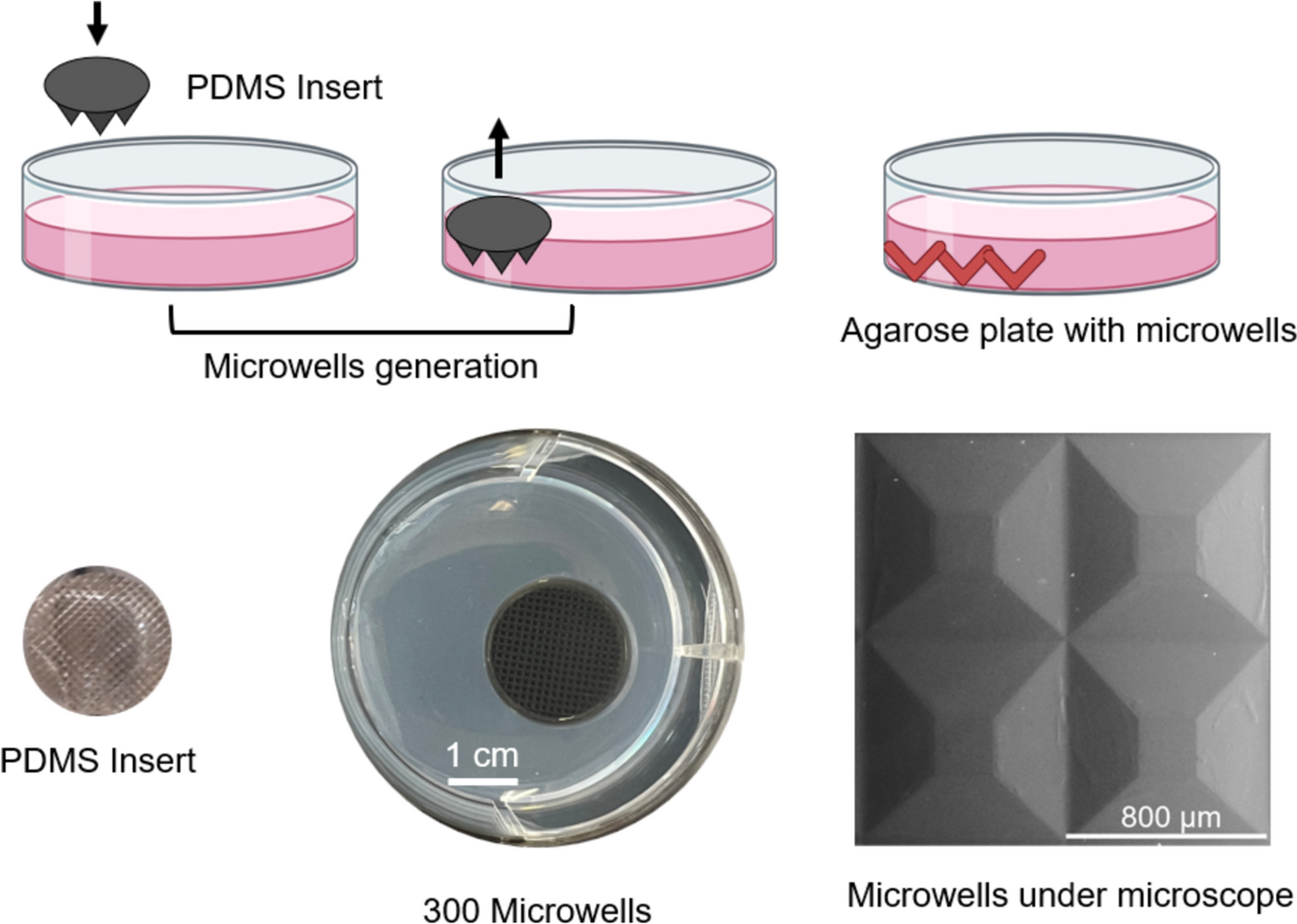
Agarose plate and microwells manufacturing and sterilisationTo create non-adherent agarose microwell plates, mold-replication technology was used as previously described (Ghezelayagh et al. 2022). To produce the microwell plates, 3 mL melted 2% agarose (50004, Lonza) was poured in a well from a 6-well plate and with the help of a polydimethylsiloxane (PDMS) insert 300 pyramidal micro-wells, each with an 800 µm diameter were stamped. After the agarose solidified, the insert was gently removed, revealing a mirror-inverted patterned agarose surface within each well (Fig. 1). The plates were sterilized by an hour of exposure to ultraviolet light before use (Zahmatkesh et al. 2022).
Fig. 1
Agarose microwells generation process and their characteristics
Cell seedingBone cells co-cultureFor the 2D bone cells co-culture model, THP-1 cells (2.4 × 104 cells per well) were seeded in the 96-well plate with 200 nM of phorbol 12-myristate 13-acetate (PMA; Cay10008014-1, Biomol) in 100 µL of THP-1 cells growing medium. After 24 h, the THP-1 cell growing medium was removed, and SCP-1 cells (0.3 × 104 cells per well) were seeded into the same well with 100 µL bone differentiation medium (50:50 mix of RPMI and MEM α, 2% FCS, 200 µM L-ascorbic acid 2-phosphate (A8960-5G, Sigma), 5 mM β-glycerolphosphate (G9422-10, Sigma), 25 mM HEPES (HN78.2, Carl Roth), 1.5 mM CaCl2 and 5 µM cholecalciferol (95,230, Sigma). The bone co-culture system was cultured in the humidified conditions incubator at 37 °C with 5% CO2 and the bone differentiation medium was replaced twice a week.
For the 3D bone cell co-culture model, after sterile control, the THP-1 growing medium was totally aspirated from the scaffolds. A single scaffold was placed at the centre of each well in a 48-well plate. Following a previously described method (Weng et al. 2020), THP-1 cells (8 × 104 cells/15 µL per scaffold) were seeded with 200 nM of PMA into the scaffold. Following 4 h of incubation at 37 °C, 5% CO2 humidified atmosphere, 500 µL THP-1 growing medium with 200 nM PMA was carefully added to each scaffold. The specimens were incubated in a humidified atmosphere with 5% CO2 at 37 °C for 24 h to ensure complete adherence. The following day, the THP-1 cell growing medium was removed before seeding. SCP-1 cells (1 × 104 cells/15 µL per scaffold) were seeded into the same scaffold. After 4 h, 500 μL of bone differentiation medium was gently added (Haussling et al. 2021). The bone co-culture scaffolds were cultured in the humidified conditions incubator at 37 °C with 5% CO2 and the bone differentiation medium was replaced twice a week.
HepaRG cell cultureFor the 2D liver model, HepaRG cells (0.9 × 104 cells per well) with 100 µL HepaRG cell culture medium were seeded into the 96-well plate. After 2 weeks of culture, cell differentiation was induced with 1.7% of DMSO for 2 weeks before the cells were used in the experiment. Cells were cultured in a humidified incubator at 37 °C with 5% CO2 and the medium was changed three times per week.
For the liver spheroids model, before seeding the HepaRG cells, we added 2 mL HepaRG cell culture medium into each agarose well to create suitable conditions for cells and centrifugation. Then, HepaRG cells (30 × 104 cells/mL per well, 1000 cells/microwell) were seeded into the wells to generate spheroids. Afterward, the plate was centrifuged (3 min, 1200 rpm) to make the cells distribute equally in the microwells. HepaRG cell distribution in the microwells was checked by light microscopy. The following day, cell differentiation was induced with 1.7% DMSO, and cells were cultured in the humidified incubator at 37 °C with 5% CO2, and the HepaRG cell culture medium was changed three times per week.
Investigating the influence of supplements contained in the bone and liver medium on the 2D bone and liver cells respectivelyTo evaluate the effect of supplements, contained in bone differentiation medium, on HepaRG cells, we treated HepaRG cells in 96-well culture plates with 25 mM HEPES, 1.5 mM Calcium chloride, 20 ng/mL Cholecalciferol, 200 µM L-Ascorbic acid 2-phosphate, and 5 mM β-Glycerolphosphate dissolved in HepaRG cell differentiation medium. Stimulation and medium change were done three times per week. Cell viability and function were measured on days 7, 14 and 21.
To evaluate the effect of supplements, contained in the HepaRG cell differentiation medium, on SCP-1 and THP-1 bone co-culture system, we seeded THP-1 and SCP-1 cells in 96-well culture plates (according to Sect. Bone cells co-culture). The following day the co-culture system was stimulated with 50 µM Hydrocortisone, 2 mM L-Glutamine, 1,7% DMSO, and 5 µg/ml Insulin dissolved in the bone differentiation medium. Stimulation and medium change were done twice a week. Cell viability and function were measured on days 7, 14 and 21.
Optimisation of culture medium for 3D liver-bone co-culture systemTo determine the optimal medium ratio that can support the viability and function of liver and bone cells, various ratios of HepaRG cell differentiation medium and bone differentiation medium (abbreviated in the text from now on as L-B medium or B-L medium) were tested for their effects on the viability and function of both compartments independently. 300 HepaRG spheroids or 2 bone scaffolds were cultivated in 100:0, 75:25, 50:50, and 25:75 ratios of L-B medium or B-L medium respectively. On days 7, 14, and 21, cell viability and function were measured compared to the control group of 100% HepaRG or bone differentiation medium respectively.
Establishment of 3D liver-bone co-culture modelOnce the spheroids were generated, the pockets for the bone scaffolds were generated by piling the cell-free agarose. Scaffolds containing bone cells were placed in the agarose pocket (Fig. 2). The ratio between bone scaffolds and HepaRG spheroids was 2:300. After combining the scaffolds with the HepaRG spheroids, HepaRG cell differentiation medium and bone differentiation medium were used to maintain the co-culture system for 21 days. Cells were cultured in a humidified incubator at 37 °C with 5% CO2 and the medium was changed three times per week.
Fig. 2
The setup of Liver spheroids-bone scaffolds co-culture system. After HepaRG spheroids were generated, the insert was used to make pockets for bone scaffolds on the agarose plate. Scaffolds containing bone cells were placed in the agarose pocket which combined with HepaRG spheroids to establish the 3D liver-bone co-culture model
Stimulation of diclofenac on the 3D liver-bone co-culture modelDiclofenac (D6899, Sigma) was dissolved in 99% formaldehyde to prepare a 10 mM stock solution, which was stored in a − 20 °C refrigerator. Based on clinical research findings, in vitro liver-bone model was treated with 3–6 µM diclofenac daily; those diclofenac concentrations represent the plasma concentration of diclofenac in humans after therapeutic use (oral intake 50 mg) (Cuklev et al. 2011; Miyatake et al. 2009; Scallion and Moore 2009).
Stimulation of 4’-hydroxydiclofenac on the 3D bone co-culture model4′-hydroxydiclofenac (32,412, Sigma; 4-OH diclofenac) was dissolved in 99% formaldehyde to prepare a 10 mM stock solution, which was stored in a − 20 °C refrigerator. Based on the amount of 4-OH diclofenac produced in our liver-bone model and the plasma concentration of 4-OH diclofenac after diclofenac use in humans. 75 nM, 300 nM, and 600 nM 4′-hydroxydiclofenac were choosed to exposure to 3D bone model daily (Degen et al. 1988; Yasar et al. 2001).
Stimulation of Interleukin-6 on the 3D bone co-culture model5 µg Interleukin-6 (200-06, Peprotech; IL-6) was first dissolved in 50 µl ddH2O, and then 450 µl 0.1% bovine serum albumin was added to prepare a 10 µg/ml stock solution, which was stored in a − 80 °C refrigerator. According to studies, 5–10 pg/ml IL-6 daily stimulation on a 3D bone model was used to mimic a human inflammatory condition. (Bakker et al. 2014; Singh et al. 2015).
Mitochondrial activity assay by resazurin conversionMitochondrial activity for bone and liver cells were evaluated after the 7th, 14th and 21st days by resazurin conversion. Bone scaffolds were washed with PBS and transferred to a new 48-well plate. Next, scaffolds were immediately covered with 500 µL of a 0.0025% resazurin solution (in PBS). Scaffolds without cells were used as background controls. After incubation for 2 h at 37 °C, the fluorescence of the produced resorufin was measured using the Omega Plate Reader (BMG Labtech, Ortenberg, Germany) at excitation/emission (ex/em) 544 nm/590–10 nm. To measure the fluorescence, 3 × 100 µL of each sample (scaffold) was transferred to the wells of a 96-well plate (Haussling et al. 2019).
HepaRG spheroids were carefully collected from agarose microwells. Before measurement, the medium was aspirated and the spheroids were washed three times with HepaRG plain medium. 200 µL of the 0.0025% resazurin solution (in plain medium) was used for 300 HepaRG spheroids. 2 × 100 µL of HepaRG spheroids in resazurin working solution were transferred to a 96-well plate, as background 100 µL resazurin working solution without incubation with cells was used (McMillian et al. 2002). The produced resorufin was measured by the fluorescence at ex/em 544 nm/590–10 nm using the Omega Plate Reader after incubation for 30 min, 60 min, 90 min, and 120 min. Mitochondrial activity was calculated by the rate of conversion of resazurin to resorufin.
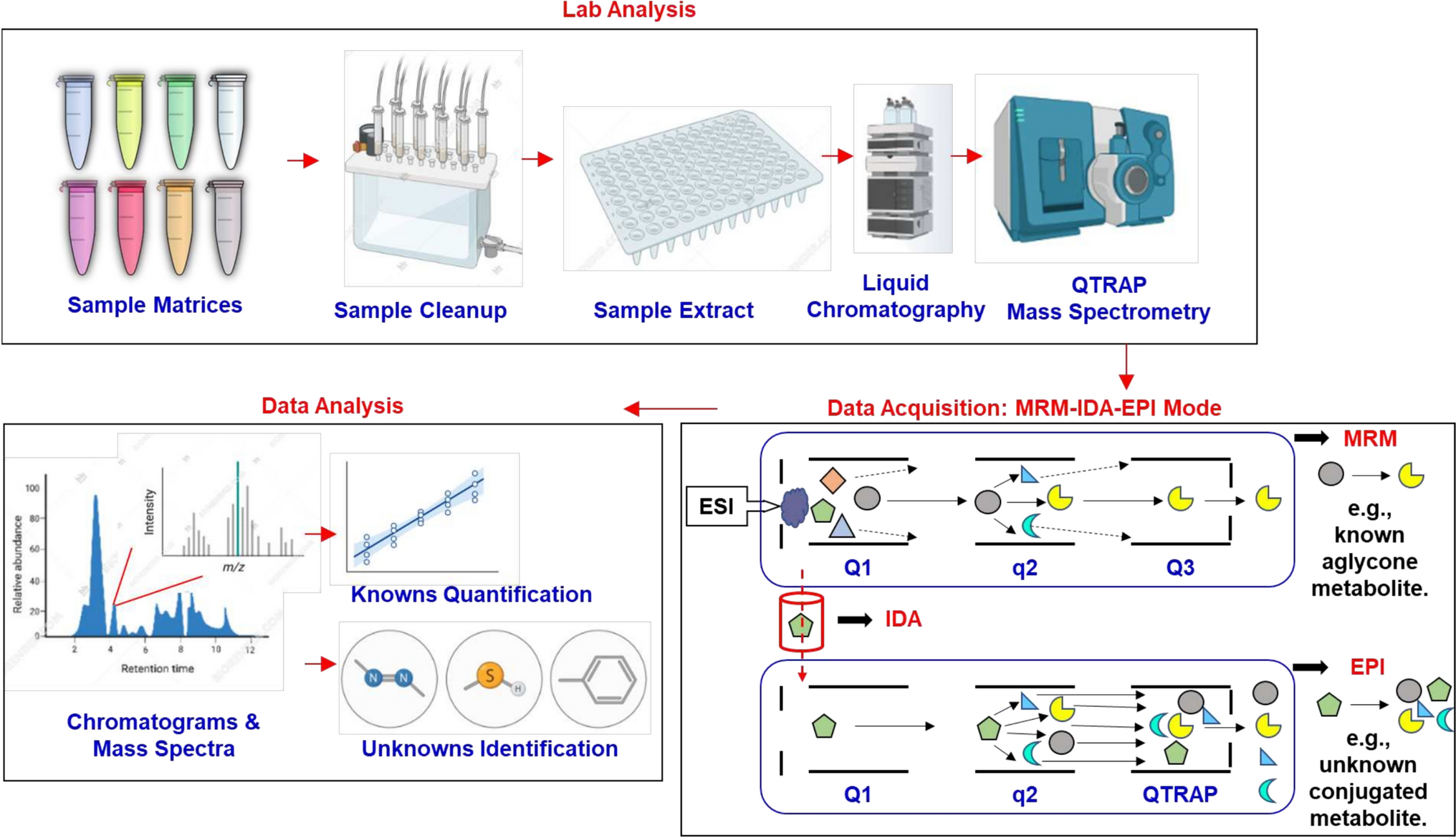
CYPs activity assay by LC-HPLC/MS-based method (Phase I enzymes)As described before (Hoffmann et al. 2012; Ruoss et al. 2019), CYP activities were measured based on the quantification of CYP-isoform-specific metabolites of different drugs using LC-HPLC/MS detection. Table 1 summarizes the selected substrates, working concentrations, incubation times, and the measured metabolites. 10 mM Phenacetin (77,440, Sigma), 10 mM Bupropion Hydrochloride (B689625, Toronto Research Chemicals), 10 mM diclofenac and 10 mM Testosterone (T1500, Sigma) stock solutions were diluted with HepaRG plain medium to a 1:1000 ratio and prepared as substrates for cocktail C1. 10 mM S-Mephenytoin (M225000, Toronto Research Chemicals) and 10 mM Bufuralol Hydrochloride (B689540, Toronto Research Chemicals) stock solutions were diluted with HepaRG plain medium to a 1:1000 ratio and prepared as substrates for cocktail C2. The HepaRG spheroids were incubated with 100 µL of working solution for C1 or C2 according to the described incubation time in Table 1. After incubation, the collected supernatants were stored at − 80 °C until measurement. Supernatants from 3 independent experiments were pooled into one sample and the CYP-isoform-specific metabolites of the sample were measured by the company Pharmacelsus (Saarbrücken, Germany) using an LC-HPLC/MS-based method. DNA content from liver spheroids was used to normalize data.
Table 1 CYPs activity by LC-HPLC/MS-based method (Phase I enzymes). Conditions, Substrates, concentrations, and measured reactionsCYP2C9 activity assay by fluorescence-based methods (Phase I enzymes)Phase I CYP2C9 enzyme activity was measured as previously described (Donato et al. 2015). Before measurement, the medium was aspirated, the HepaRG spheroids were washed three times with the HepaRG plain medium and 200 µL of the 5 µM Dibenzylfluoresceine and 10 µM 3,3′-Methylene-bis(4-hydroxycoumarin) (working solution—freshly prepared in HepaRG plain medium) were added to each sample of HepaRG spheroids. HepaRG spheroids in 100 µL with the working solution were transferred to a well of a 96-well plate in duplicates, and for the negative control, 100 µL of the working solution was transferred into a well without spheroids. The produced fluoresceine was measured by fluorescence at ex/em 485 nm/520 nm using the Omega Plate Reader after incubation for 30 min, 60 min, 90 min, and 120 min. CYP2C9 enzyme activity was calculated by the rate of Dibenzylfluoresceine to fluoresceine. DNA levels from liver spheroids were used to normalize data.
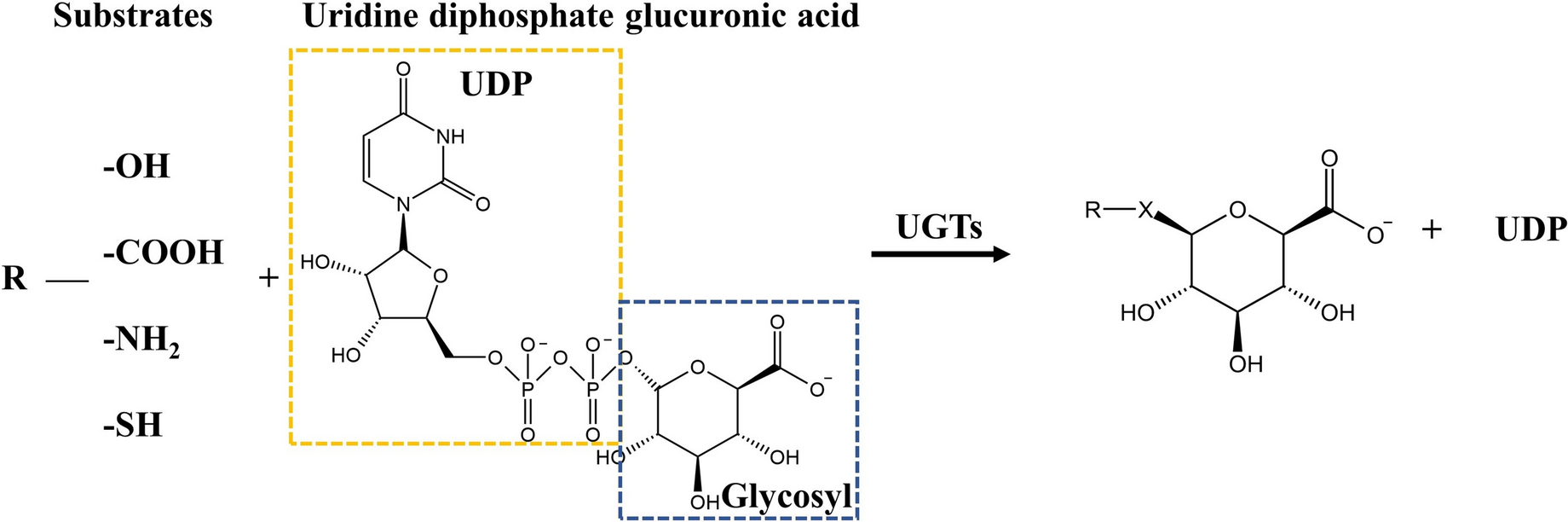
UGT activity by fluorescence-based methods (Phase II enzymes)An adjusted method previously reported was utilized to measure the activities of Phase II enzymes (Hammour et al. 2022). 200 µL of the 6.25 µM 4-Methylumbelliferone (in HepaRG plain medium) was used for 300 HepaRG spheroids. 100 µL HepaRG spheroids with the working solution were transferred to a 96-well plate to generate duplicates, and for the negative control, 100 µL of the working solution was transferred into a well without cells. The fluorescence of 4-Methylumbelliferone was measured at ex/em 355 nm/460 nm using the Omega Plate Reader after incubation for 30 min, 60 min, 90 min, and 120 min. DNA concentration from liver spheroids was used to normalize data.
Alkaline phosphatase (AP) activity assay by absorbance-based methodAs an initial indicator of osteogenesis, the activity of alkaline phosphatase (AP) was assessed (Aspera-Werz et al. 2018). The scaffolds were washed three times with PBS and then incubated with 500 µL of AP reaction buffer (3.5 mM 4-nitrophenyl-phosphate, 50 mM glycine, 1 mM MgCl2, and 100 mM TRIS; pH 10.5) for a period of 2 h at 37 °C to allow for reaction. The photometric quantification of the conversion of 4-nitrophenyl-phosphate to 4-nitrophenol in 100 µL solution in triplicates was carried out (λ = 405 nm; Omega Plate Reader). The obtained experimental values were adjusted based on the background control, which was a scaffold without cells (Guo et al. 2022). Following this, the data was normalized to DNA concentration.
Carbonic anhydrase (CA II) activity by absorbance-based methodsAs an early indicator of osteoclast differentiation, CA II was assessed (Zhu et al. 2020). The cells/scaffolds were washed three times with PBS and then incubated with 100/500 µL of CA II reaction buffer (12.5 mM TRIS, 75 mM of NaCl, and 200 mM of 4-nitrophenyl acetate; pH 7.5) for 15 min at 37 °C. CAII activity was calculated by the rate of conversion of 4-nitrophenyl acetate to 4-nitrophenol. The resulting reaction’s product, 4-nitrophenol, was quantified using a photometer (λ = 405 nm; Omega Plate Reader). The obtained experimental values were adjusted based on the background control, which was a scaffold without cells (Guo et al. 2022). After that, the data was normalized to DNA concentration.
Tartrate-resistant acid phosphatase (TRAP) activity by absorbance-based methodsA late marker for osteoclast activity, TRAP was assessed (Zhu et al. 2020). 30 µL of pooled supernatant was mixed with 90 µL of TRAP activity assay solution (5 mM 4-nitrophenyl phosphate, 100 mM sodium acetate, and 50 mM sodium tartrate in demineralized water; pH 5.5) to react for 6 h at 37 °C. To terminate the reaction, 90 µL/well of 1 M NaOH was added, effectively suppressing further reaction. The resulting reaction product, 4-nitrophenol, was quantified using a photometer (λ = 405 nm; Omega Plate Reader). The obtained experimental values were adjusted based on the background control, which was a scaffold without cells (Guo et al. 2022). After that, data was normalized to DNA concentration.
Alizarin red stainingTo assess matrix mineralization, cells were fixed with 100% ethanol overnight at − 20 °C. The fixed cells were gently washed with tap water, stained with 50 µL of 0.5% w/v Alizarin Red solution (pH 4.0), and incubated at room temperature for 30 min protected from light. Excess Alizarin Red staining solution was removed by washing the cells with tap water. For quantitative measurement, the bound Alizarin Red was dissolved with 10% w/v Cetylpyridinium chloride solution and quantified photometrically (λ = 562 nm; Omega plate reader) as described previously (Zhu et al. 2020).
Sulforhodamine B (SRB) stainingSRB staining was performed to measure the total protein content of samples. Cells were directly fixed by ethanol for at least 60 min at − 20 °C after washing with PBS. Next, plates were incubated with 0.4% w/v SRB for 30 min at RT in the darkness. Then the samples were washed 4–5 times with 1% v/v acetic acid to remove unbound SRB. Afterwards, bound SRB was resolved with 10 mmol/L unbuffered TRIS solution (pH 10.5). SRB staining results were determined by absorbance (λ = 565 nm) with a plate reader (Ehnert et al. 2018).
DNA isolation and quantificationAs previously described (Haussling et al. 2019), DNA measurement was performed to assess cell viability and data normalisation. For the HepaRG spheroids, spheroids were collected from agarose microwell plates, washed with PBS, and then added to 100 µL of hot (98 °C) 50 mM NaOH solution and incubated at 98 °C for 5 min. After vortexing, the tubes containing the HepaRG spheroids and NaOH were frozen at − 20 °C. The following day, 110 μL TRIS (0.1 M, pH = 8.0) was added to all thawed samples and centrifuged at 20,000 × g for 10 min. For the bone scaffolds, scaffolds were transferred from agarose plates to a new 48-well plate. 250 µL of hot (98 °C) 50 mM NaOH solution was added to each scaffold. After 5 min of incubation, the scaffolds with NaOH were frozen at − 20 °C. The following day, 275 μL TRIS (0.1 M, pH = 8.0) was added to all thawed samples. After fully mixed, the liquid was collected and centrifuged at 20,000 × g for 10 min. For the absorption-based quantification, all samples were measured on the LVIS Plate (BMG Labtech, Ortenberg, Germany) in two steps. For the first step, 2 µL of DNA isolation buffer (500 µL NaOH (50 mM), and 550 µL TRIS (0.1 M, pH8.0)) as a blank was measured. In the second step, 2 µL of each DNA sample was measured in duplicates. Then, DNA concentrations were calculated at λ = 260 nm by the Omega analyzation software MARS.
Mineral content of bone scaffoldThe mineral content of the scaffold was analysed by performing quantitative computer tomographic (CT) scans utilizing a clinical CT scanner with 128 slice capability (SOMATOM Definition Edge, Siemens Healthineers, Erlangen, Germany). The specimens underwent scanning using the following parameters: an 80 kV tube voltage, an effective tube current of 500 mAs, an acquisition of 16 × 0.3 mm with a slice thickness of 0.4 mm, and a pitch of 0.4. Subsequently, the images were reconstructed using an advanced very sharp reconstruction kernel (V80u) with iterative image reconstruction technology (SAFIRE, Siemens Healthineers, Erlangen, Germany) with grade 5 and displayed in a bone window. Rectangular axial reconstruction field-of-view was about 10 cm x 10 cm with matrix resolution of 512 × 512 pixel. Obtained DICOM images were imported into the ImageJ software using the “DICOM sort” plugin. The resulting stack was cropped to show the area of interest. From each scaffold, the mean integrated density was determined and normalized to the reference block (Phantom EFP-06-96) (Weng et al. 2020).
Stiffness of bone scaffoldThe stiffness of the bone scaffold was measured using Young’s modulus as described previously (Weng et al. 2020). A ZwickiLine Z 2.5TN machine (Zwick GmbH and Co. KG, Ulm, Germany) vertically squeezed the surface scaffolds with the speed of 5 mm/min up to uniaxially by 10% of the original high of the scaffold. A force sensor recorded the applied force in real time. The stiffness of the scaffold was calculated:
$$Young^s \;modulus\left[ \right] = \frac \right]}} } \right] \times change \;in \;height \left[ \right]}}$$
Dot blot analysisDot blot was used to detect secreted protein in the culture supernatant, following the previously described method (Guo et al. 2022). 60 or 100 µL of supernatant was transferred onto a wet nitrocellulose membrane with a vacuum pump in a 96-well dot blotter (Carl Roth). After blocking them with 5% bovine serum albumin in TRIS-buffered saline/Tween-20 (TBS-T) for 1 h, membranes were incubated with primary antibodies at 4 °C overnight. After washing them with TBS-T, the membranes were incubated with the respective secondary antibody for 2 h. The antibodies used are summarized in Table 2. The signals were detected by chemiluminescence with a mixture containing 100 mM TRIS (T1503, Sigma), 1.25 mM Luminol (4203.1, Carl Roth), 0.2 mM p-coumaric acid (9908.1, Carl Roth), and 0.03 v/v H2O2 (CP26.5, Carl Roth), and quantified by ImageJ software.
Table 2 The antibodies used for dot blotDetection of reactive oxygen species (ROS) by 2′,7′-dichlorofluorescein-diacetate (DCFH-DA) assayHepaRG spheroids were washed three times with HepaRG plain medium before measurement. 100 µL of the 10 µM DCFH-DA working solution was used for HepaRG spheroids. After 30 min at 37 °C incubation, spheroids were washed by PBS three times. The spheroids were stimulated with diclofenac according to the setup of the experiment, and 0.03% H2O2 was used to stimulate spheroids as a positive control. The produced 2′7′-dichlorofluorescein (DCF) was continuously measured by the fluorescence at ex/em 485 nm/520 nm using the Omega Plate Reader during 0–10 min (Aspera-Werz et al. 2018).
Detection of reduced glutathione (GSH) and oxidized glutathione (GSSG) by Ellman assayAfter stimulation with 3–6 µM diclofenac, HepaRG spheroids were collected and washed three times with cold PBS before measurement. 3% W/V m-phosphoric acid was used to lysate cells to precipitate the proteins. Protein samples were re-neutralized with 5 mM EDTA in 0.1 M potassium phosphate buffer. After that, samples were centrifuged at 3000 × g for 10 min and then the supernatant was collected for GSH and total GSH measurements. For the determination of GSH, 20 µl of the sample was mixed with 120 µl of 0.56 mM 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) in 0.1 M potassium phosphate buffer. To determine total GSH, 20 μl of the sample was incubated for 30 s with 120 μl of a mixture (1:1) of 1.68 mM DTNB and 2.5 U/ml glutathione reductase in 0.1 M potassium phosphate buffer. Then, 60 μl of NADPH 0.8 mM was added and absorbance was measured at λ = 412 nm. GSSG is obtained by subtracting GSH from the total GSH (Aspera-Werz et al. 2018).
Statistical analysesThe data are presented as means ± the standard error of the mean (SEM). All the experiments were repeated at least three times with two to four technical replicates. Statistical analyses were performed using GraphPad Prism software (GraphPad Software 9.0, La Jolla, CA, USA). The data of the two groups were compared with the Mann–Whitney test. The data of multiple groups were compared with the non-parametric Kruskal–Wallis test, followed by Dunn’s multiple comparison test. A two-way ANOVA test followed by Turkey’s multiple comparisons was used when two independent variables were compared among groups. A p-value of < 0.05 was considered statistically significant.
留言 (0)