
記住我




Adult male Wistar rats (280–380 g; n = 15) were housed in standard cages with ad libitum access to food and water under controlled laboratory conditions (21 ± 1 °C; 40–60% humidity; 12:12 day/night cycle, lights on at 8:00 AM). All experimental procedures were approved by the Boğaziçi University Institutional Ethics Committee for the Use of Animals in Experiments (BÜHADYEK) and carried out by licensed personnel.
Stereotaxic surgery and retrograde tract-tracingAnimals were anesthetized with either intraperitoneal (IP) injections of ketamine (80 mg/kg)—xylazine (13.3 mg/kg) (retrograde tracing experiments) or isoflurane (4% for induction, 1–2% for maintenance). Following induction of anesthesia, a local anesthetic (Vemcaine, 10%) and a povidone-iodine solution were applied to the shaved forehead before placing the animal in the stereotaxic frame (Kopf Instruments, USA). A homeothermic heating pad was used to monitor and maintain the body temperature at 36 °C. Craniotomies were performed above the anterior–posterior (AP) and medial–lateral (ML) coordinates of the target nuclei.
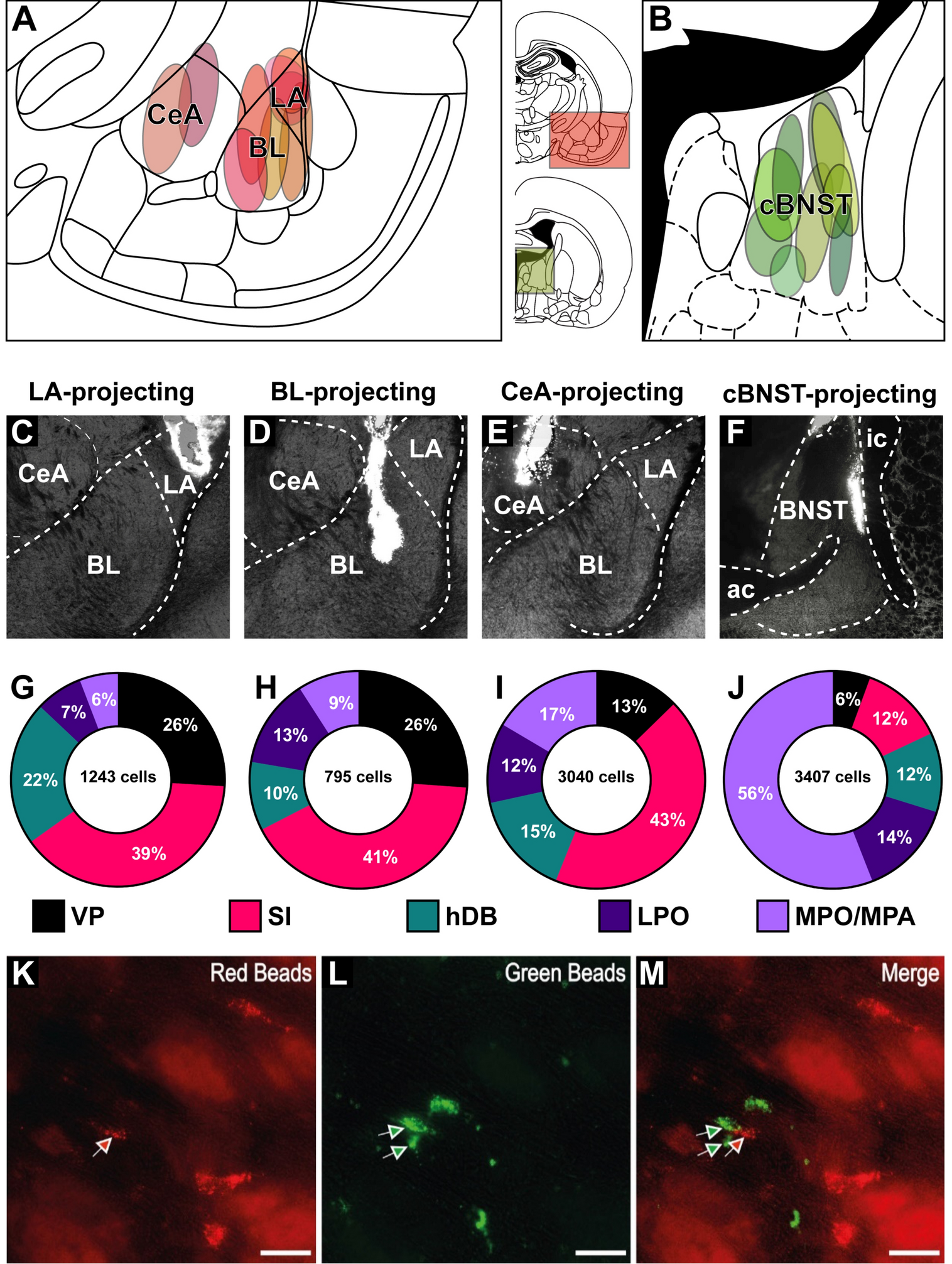
For retrograde tracing, red (diluted in saline by 1:2, volume = 200 nl) and green (undiluted, volume = 200 nl) fluorescent latex microspheres (Retrobeads, Lumafluor Inc., USA) were injected into the LA (AP = −2.80, ML = ± 5.30, DV = −7.30), BL (AP = −2.80, ML = ± 4.60, DV = −8.20), CeA (AP = −2.40, ML = ± 4.20, DV = −8.00), and the central BNST (cBNST) (AP = −0.48, ML = ± 1.40, DV = −6.00) (Paxinos and Watson 2007); Fig. 1A–B). In each animal, we injected one color of Retrobeads into one of the amygdaloid nuclei (LA, BL or CeA), while the other color of tracer was injected into the cBNST in the same hemisphere. Injections were performed unilaterally. The right and left target hemispheres were counterbalanced. The tracers (green or red Retrobeads) were also counterbalanced for each target region.
Fig. 1
Retrobeads injections and resulting retrograde labeling in the basal forebrain and preoptic nuclei. A, B Injection sites targeting the amygdala nuclei (A) and the cBNST (B). C–F Brightfield photographs of representative injections in the LA (C), BL (D), CeA (E) and cBNST (F). G–J Donut charts demonstrating the percentage of LA (G), BL (H), CeA (I) and cBNST (J) targeting neurons in the observed basal forebrain and preoptic nuclei. K–M Fluorescent micrographs of retrogradely labeled neurons with red (K) or green (L) Retrobeads. ac anterior commissure, BL basolateral amygdala, cBNST central bed nucleus of stria terminalis, CeA central amygdala, hDB nucleus of horizontal limb of the diagonal band of Broca, ic internal capsule, LA lateral amygdala, LPO lateral preoptic area, MPO/MPA medial preoptic nucleus/area, SI substantia innominata, VP ventral pallidum
For anterograde tracing, AAV9-CaMKIIα-hM4D(Gi)-mCherry (250 nl, 1 × 1013 vg/ml, Addgene plasmid # 50,477, bilateral injection, n = 4) or AAV1-hSyn-EGFP-cre (250 nl, 1 × 1013 vg/ml, Addgene plasmid #105,540, unilateral injection, n = 2) were injected into the VP (AP = −0.20, ML = ± 2.5, DV = −7.80) or SI (AP = −1.00, ML = ± 2.50, DV = −8.10), (Paxinos and Watson 2007); Fig. 4A–B). AAV serotype 1 (AAV1) causes robust anterograde transsynaptic expression, enabling visualization of downstream synaptic targets of regionally specified starter cells (Zingg et al. 2022). Here, we injected AAV1-hSyn-EGFP-cre into the VP to transsynaptically label downstream neurons in the amygdala.
All injections were made with a microinjection syringe pump and a 1 μL micro-injection syringe (Hamilton, NV, USA). Each tracer injection took 5 min (40 nl/m for Retrobeads and 50 nl/m for viral tracing), after which the syringe was maintained at the target location for 10 min before retrieval to minimize dorsal diffusion. Once the incision was sutured, a local analgesic (Anestol pomade, 5% lidocaine and Jetokain, 5 mg/kg) was applied to the cranial surface before the animal was removed from the stereotaxic apparatus. The animals underwent a 5-day post-surgical recovery period in order to ensure maximal axonal transport of the Retrobeads or a 14-day post-surgical recovery period to allow optimal viral-driven expression of fluorescent proteins.
Perfusion-fixation and tissue preparationFollowing the recovery period, animals were deeply anesthetized with the ketamine (80 mg/kg)—xylazine (13.3 mg/kg) mixture (IP), and perfused transcardially with 0.9% saline and 4% depolymerized paraformaldehyde (PFA) in 0.1 M PBS. Removed brains were post-fixed in the same fixative solution for 24–48 h at 4 °C. They were thoroughly rinsed following post-fixation and transferred to 0.1 M PB for slicing. Serial 60–80 µm thick coronal sections were obtained using a Leica VT 1000S vibratome (Leica Microsystems, Germany).
ImmunohistochemistryWe conducted immunofluorescence labeling as described previously (Unal et al. 2015b; Akmese et al. 2023; Kingir et al. 2023). PBS with 0.3% Triton X-100 (PBS-TX) was used in all solutions and rinsing procedures to achieve tissue penetration. Coronal sections were rinsed 3 times (10 min each) in PBS-TX, followed by 1 h blocking at room temperature (RT) in 20% Normal Horse Serum (NHS) or Normal Goat Serum (NGS), depending on the secondary antibody. The sections were then incubated for 72 h at 4 °C in PBS-TX containing the primary antibodies and 1% NHS/NGS (refer to Table 1 for primary antibodies). Following 3 × 10 min of rinsing, the sections were incubated in the secondary antibody solution containing 1% NHS/NGS in PBS-TX for 4 h at RT. Sections were subsequently mounted and cover-slipped using methyl salicylate (Sigma-Aldrich, MO, USA) and examined using an epifluorescence (Olympus BX43) or confocal microscope (Leica SP8, Leica Microsystems).
Table 1 Primary antibodiesRetrogradely labeled neurons were tested for different molecules listed in Table 1. Immunoreactivity for PV, CB, or AT-rich sequence-binding protein-1 (SATB1) was examined to identify non-cholinergic, putative GABAergic or glutamatergic neurons. ChAT immunoreactivity was assessed to identify cholinergic neurons, and Leu-enkephalin was used as a regional marker for VP (Fig. 1B). We used the following secondary antibodies: goat anti-rabbit Alexa Fluor 405 (1:250; A31556, Invitrogen), donkey anti-rabbit Alexa Fluor 488 (1:250; ab150073; Abcam), donkey anti-goat Cy3 (1:250; 705-165-147; Jackson ImmunoResearch Laboratories), donkey anti-goat DyLight650 (1:1000, ab96938, Abcam).
Sections from each brain were stained with DAPI or cresyl violet to facilitate histological identification of injection sites and cytological differentiation of the BF nuclei. DAPI staining was performed by incubating sections in the DAPI solution (1:2000, D3571, ThermoFisher) for 10 min. The sections were rinsed in PBS 3 times (10 min each) at RT. For cresyl violet staining, the sections were mounted on slides 3 days before the procedure and incubated for 1 min at 40 °C immediately before the staining. Slides were transferred through 100% ethanol (EtOH) (2 min), two sets of xylenes (2 min each), 100% EtOH (2 min), 70% EtOH (2 min), 20% EtOH (2 min), dH2O (1 min), cresyl violet solution (0.5 g cresyl violet acetate and 15 ml acetic acid in 500 ml dH2O, 15 min), differentiation solution containing 70% EtOH and 10% acetic acid (10 s), differentiation solution containing 100% EtOH and 10% acetic acid (10 s), 100% EtOH (5 min) and two set of xylenes (5 min each). Slides were then cover-slipped using Entellan new (Merck) mounting medium and examined under a light microscope.
MicroscopyThe initial observations were conducted utilizing Olympus cellSens Imaging Software v2.2 on an epifluorescence microscope (Olympus BX43) equipped with a monochrome CCD camera (Olympus XM10). The images were obtained with 4x (Plan Apochromat, N.A. = 0.02, Nikon), 10x (Plan Fluor, N.A. = 0.30, Nikon), and 20x (Plan Fluor, N.A. = 0.50, Nikon) objective lenses. The 4 × objective lens was used for histological analysis, and 10 × and 20 × objective lenses were used to locate retrogradely labeled neurons in the target basal forebrain nuclei. Four fluorescent filter sets (for DAPI, Alexa Fluor 488, Cy3, and Cy5) were used for the detection of Alexa Fluor 405 fluorophores and DAPI, Alexa Fluor 488 fluorophores and green Retrobeads, Cy3 fluorophores and red Retrobeads, and DyLight 650 fluorophores respectively.
Following wide-field microscopic observations, multichannel fluorescence images were acquired with a Leica SP8 confocal microscope (Leica Microsystems, Wetzlar, Germany) using the LAS X software (Leica Microsystems) at a minimum pixel resolution of 1024 × 1024. The images were obtained with 20x (Plan Fluotar, N.A. = 0.4, dry, Leica Microsystems) or 40x (Plan Apochromat, N.A. = 1.10, water-immersion, Leica Microsystems) objective lenses. We employed four distinct lasers with wavelengths of 405, 488, 552, and 638 nm, along with PMT or HyD sensors, to visualize the fluorescence signal. The pinhole size was configured to 1 Airy unit. In the acquisition of z-stacks, the z-stack step size was set at half the optical section thickness. Post-acquisition brightness and contrast adjustments were performed uniformly on the whole image using the “FIJI” distribution of the ImageJ software (Schindelin et al. 2012). No non-linear or selective image adjustments were made on the acquired images.
Anatomical quantificationRetrogradely labeled neurons were manually counted in every other coronal section containing the labeled basal forebrain or preoptic nuclei. Observations and counting were made in the ventral pallidum, substantia innominata, horizontal diagonal band (hDB), lateral preoptic nucleus (LPO), and hypothalamic medial preoptic nucleus/area (MPO/MPA). Neuron quantification in the rostral-most parts of the extended amygdala was included in the SI. Cell counts were added together to obtain a total labeled neuron value for each region of interest. Normalized counts were derived by dividing the total number of labeled somata quantified in each nucleus by the number of observed sections. A similar quantification method was followed for the immunolabeled neurons. For each section, we counted all the cell bodies that were immunoreactive for a molecular marker in each region and noted the number of neurons that showed colocalization with Retrobeads. For each BF nucleus, we then calculated the percentage of neurons expressing each tested molecular marker among the observed retrogradely labeled neurons projecting to the LA, BL, CeA or the cBNST. Drawings depicting the distribution of labeled neurons in the BF were made with a camera lucida. All figures were created using Adobe Illustrator (v 25.0).
We analyzed the normalized fluorescence intensity of anterogradely labeled axonal fibers using the FIJI distribution of ImageJ software (Schindelin et al. 2012). Specifically, we measured the fluorescence intensity of VP/SI axonal fibers within the LA, BL, the lateral CeA (encompassing the capsular [CeAc] and lateral [CeAl] subdivisions), and the medial CeA (CeAm) in both hemispheres. Fluorescence intensity is reported in arbitrary units (AU). Data from both hemispheres were pooled for statistical analyses. To compare the normalized density of VP/SI axonal fibers across amygdala subnuclei, we performed a Student’s t-test.
The density of transsynaptically labeled neurons was quantified along the rostrocaudal axis in serial sections spaced 250 μm apart. Neuronal counts were conducted in the LA, BL, the lateral division of the CeA (CeAc and CeAl), and CeAm. Linear regression analysis was used to evaluate changes in neuronal density along the rostrocaudal axis.
Model implementationBLA network modelA 1000-neuron network model of the BLA was developed to investigate the potential role of the dense non-cholinergic, putative GABAergic projection, originating from the VP/SI and targeting the input nuclei of the basolateral amygdala, namely the LA and BL. The model incorporated our observations and previous reports on the synaptic parameters of the intrinsic (within BLA) and extrinsic (afferents to the BLA) connectivity. All parameters were derived from available rat or mouse amygdala data. The model was developed using the Allen Institute’s Brain Modeling Toolkit (BMTK) with the NEURON 7.7 simulator (Carnevale and Hines 2006), with a fixed time step of 0.1 ms. Verification of network connectivity parameters and plot generation were performed using the Python package BMTools. For the analysis of neuron power spectral density (PSD) and frequency, we computed spectrums utilizing the Welch Periodogram method (pwelch in MATLAB). Subsequent analyses and plot generation were performed using standard Python codes. The complete model is accessible for download on GitHub at https://github.com/tjbanks/AmygdalaTheta.
Single cell modelsFor computational modelling, we used the most parsimonious model that sufficiently explains the phenomena explored (Bassett et al. 2018). We modeled two types of principal neurons (PNs) and the three most populous groups of GABAergic interneuron that have been connected to local oscillations in the amygdala. The resulting network was able to reproduce relevant in vitro and in vivo properties of the BLA as described later.
The principal neurons (n = 800) were divided into two electrophysiological subtypes as Type A (adapting; PNA) (n = 569) and Type C (continuous; PNC) (n = 231). These model cells were adapted from our prior work (Feng et al. 2016, 2019). The three major groups of interneuron consist of (1) PV + Basket cells (n = 93), (2) calretinin (CR +) interneurons that often co-express vasoactive intestinal polypeptide (VIP) (Mascagni and McDonald 2003) and include interneuron-specific interneurons (Rhomberg et al. 2018) and small cholecystokinin (CCK)-expressing cells (n = 56), and (3) somatostatin (SOM)-expressing interneurons that include neurogliaform cells (NGFC; n = 51). The proportion of each neuronal group (56.9% PNA, 23.1% PNC, 9.3% PV + interneuron, 5.6% CR + interneuron, and 5.1% SOM + interneuron) was derived from previous reports (McDonald and Mascagni 2001; Mascagni and McDonald 2003; McDonald 2020). The parameters of single cell models are listed in Table 2.
Table 2 Parameters of single cell modelsPrincipal neurons had three compartments: soma (diameter 24.75 µm, length 25 µm), a proximal dendrite (diameter 3 µm; length 270 µm), and an apical dendrite (diameter 5 µm; length 555 µm) to match passive properties. The specific membrane resistance, membrane capacity, and cytoplasmic (axial) resistivity values were as follows: Rm = 40 ± 5 kΩ-cm2, Cm = 2.4 µF/cm2, and Ra = 150 Ω-cm. The leakage reversal potential (EL) was set to −75 ± 4 mV. This configuration resulted in a resting membrane potential (Vrest) of −66 ± 4 mV for both types A and C cells. The input resistance (RIN) was 140 ± 20 MΩ and 360 ± 20 MΩ, and time constant (τm) was ~ 30 ms and ~ 20 ms, for Type-C and Type-A cells, respectively. These values fell within the reported ranges observed in physiological studies (Washburn and Moises 1992). The soma and dendrite compartments had the following currents: leak (IL), voltage-gated persistent muscarinic (IM), high-voltage activated Ca2+ (ICa), spike-generating sodium (INa), potassium delayed rectifier (IDR), A-type potassium (IA) (Li et al. 2009; Power et al. 2011), and hyperpolarization-activated nonspecific cation (Ih) current. In addition, the soma exhibited a slow apamin-insensitive, voltage-independent afterhyperpolarization current (IsAHP) (Power et al. 2011; Alturki et al. 2016). The axonal compartments had the following currents: leak (IL), high-threshold sodium (INa1.2), low-threshold sodium (INa1.6), and potassium delayed rectifier (IDR) (Hu et al. 2009). PNs exhibited adaptation characteristics, modulated by the magnitude of the Ca2+-dependent K+ current, set at either 50 mS/cm2 for Type A or 0.2 mS/cm2 for Type C (Kim et al. 2013). PN models were equipped with features for low- and high-threshold oscillations, designed to closely replicate physiological parameters (Pape et al. 1998; Li et al. 2009; Kim et al. 2013; Feng et al. 2016).
The PV + interneuron model contained two compartments: a soma-axon (diameter 15 µm; length 15 µm) and a dendrite (diameter 10 µm; length 150 µm). Each compartment contained a fast Na+ (INa) and a delayed rectifier K+ (IDR) current.
The passive membrane properties of PV + interneurons were characterized by a specific membrane resistance (Rm) of 20 ± 1 kΩ-cm2, a membrane capacity (Cm) of 1 µF/cm2, and distinct cytoplasmic resistivities (Ra) for the soma (3375 Ω-cm) and dendrite (150 Ω-cm). The resulting Vrest was −70 mV, input resistance (RIN) was 371 MΩ, and time constant (τm) was 20 ms. The resulting current injection responses fell within the ranges reported in earlier reports (Faber et al. 2001; Sah et al. 2003; Rainnie et al. 2006).
The CR + and SOM + interneuron models contained three compartments: a soma-axon (diameter 10 µm; length 20 µm) and two dendrites (diameter 3 µm; length 250 µm). Each compartment contained persistent Na + (INaP), potassium delayed rectifier current (IKDR), voltage-gated persistent muscarinic current (IM), and transient sodium channel (INat). In addition, the CR + interneuron contained l-calcium current, fast Na + (INa), h channel (IH), and voltage-independent afterhyperpolarization current (IsAHP). The CR + interneuron model exhibited a membrane resistance of 80 ± 1 kΩ-cm2, membrane capacity of 1 µF/cm2, and a compartmental resistance was 150 Ω-cm. The resulting Vrest was −60 mV, input resistance (RIN) was 321 MΩ, and time constant (τm) was 20 ms, as documented in earlier work (Kawaguchi and Kubota 1996; Porter et al. 1998; Caputi et al. 2009). The passive membrane properties of the SOM + interneurons were as follows: Rm = 80 ± 1 kΩ-cm2, Cm = 1.3 µF/cm2 and Ra = 150 Ω-cm. The resulting Vrest was −70 mV, input resistance (RIN) was 290 MΩ, and time constant (τm) was 19 ms. These values were within the ranges reported for SOM-containing interneurons (Karagiannis et al. 2009; Sosulina et al. 2010; Fanselow and Connors 2010).
Intrinsic and synaptic currentsThe dynamics for each compartment (soma-axon or dendrite) followed the Hodgkin-Huxley formulation as previously described (Kim et al. 2013) in Eq. 1,
$$C_ dV_ /dt = - g_ \left( - E_ } \right) - g_ \left( - V_ } \right) - \sum I_^ \sum I_^ + I_ ,$$
(1)
where \(_/_\) are the somatic/dendritic membrane potential (mV), \(_^\) and \(_^\) are the intrinsic and synaptic currents in the soma, \(_\) is the electrode current applied to the soma, \(_\) is the membrane capacitance, \(_\) is the conductance of the leak channel, and \(_\) is the coupling conductance between the soma and the dendrite (similar term added for other dendrites connected to the soma). The intrinsic current \(_^\), was modeled as \(_^=_^^(_-_)\), where \(_\) is its maximal conductance, m its activation variable (with exponent p), h its inactivation variable (with exponent q), and \(_\) its reversal potential (a similar equation is used for the synaptic current \(_^\) but without m and h). The kinetic equation for each of the gating variables x (m or h) takes the form
$$\frac} = \frac \left( } \right]_ } \right) - x}} \left( } \right]_ } \right)}},$$
(2)
where \(_\) is the steady state gating voltage- and/or Ca2+- dependent gating variable and \(_\) is the voltage- and/or Ca2+- dependent time constant. The equation for the dendrite follows the same format with ‘s’ and ‘d’ switching positions in Eq. 1.
Excitatory transmission was mediated by AMPA and NMDA receptors, while inhibitory transmission was modeled via GABAA receptors. The corresponding ionic currents were modeled by dual exponential functions (Destexhe et al. 1994; Durstewitz et al. 2000), as shown in Eqns. 3–5,
$$\begin I_} = w \times G_} \times \left( } } \right) \hfill \\ G_} = g_} \times F_} \times s\left( V \right) \times r_} \hfill \\ r_} \begin \kern-8pt/ \quad \end = ~\alpha Tmax_} \times ON_} \times \left( } }} \right) - \beta _} \times r_} , \hfill \\ \end$$
(3)
$$\begin I_ = w \times G_ \times \left( } \right) \hfill \\ G_ = g_ \times F_ \times s\left( V \right) \times r_ \hfill \\ r_ \begin \kern-8pt/ \quad \end = \alpha Tmax_ \times ON_ \times \left( } \right) - \beta_ \times r_ , \hfill \\ \end$$
(4)
$$r_ \begin \kern-8pt/ \quad \end = \alpha Tmax_ \times ON_ \times \left( } \right) - \beta_ \times r_ ,$$
(5)
where V is the membrane potential (mV) of the postsynaptic compartment (dendrite or soma), I is the current injected into the compartment (nA), G is the synaptic conductance (µS), \(w\) is the synaptic weight (unitless), and E is the reversal potential of the synapse (mV). gx,max is the maximal conductance (µS), F implements short-term plasticity, and rx determines the synaptic current rise and decay time constants based on the terms αTmax and β (Destexhe et al. 1994). The voltage-dependent variable s(V) which implements the Mg2+ block was defined as s(V) = [1 + 0.33 exp(−0.06 V)]−1 (Zador et al. 1990). The terms ONNMDA and ONAMPA were set to 1 when the corresponding receptor was open, and 0 when it was closed. Reversal potential, rise/decay time constants, and conductance for the model were derived from previously published data (Thomson and Deuchars 1997; Galarreta and Hestrin 1997; Mahanty and Sah 1998; Porter et al. 1998; Weisskopf et al. 1999; Feng et al. 2019). Synaptic weights (w) for all connections followed a log-normal distribution with a cutoff set at three times the mean to avoid non-physiological values. The parameters for ionic currents are detailed in Table 3.
Table 3 Ionic current parametersIntrinsic connectivityThe neuronal composition of the BLA network model comprised 56.9% PNA (n = 569), 23.1% PNC (n = 231), 9.3% PV + interneurons (n = 93), 5.6% CR + interneurons (n = 56) and 5.1% SOM + interneurons (n = 51). The PNs possess mutual connections with all interneuron groups. PV + interneurons target somata of the PNs, as well as the SOM + interneurons and other PV + cells, but not the CR + group. CR + interneurons form inhibitory synapses on all other neuron groups, similar to PNs. SOM + interneurons, in contrast, only target PNs and avoid the other groups (Fig. 5A). The probability of unidirectional or reciprocal synaptic connections between PNs and interneurons was set to 16%. Axonal conduction delay was distance-dependent using a conduction velocity of 500 μm/ms. Synaptic connectivity parameters are listed in Table 4.
Table 4 Synaptic connectivity parametersExtrinsic connectivityThe network model integrates thalamic/cortical glutamatergic afferents to the BLA, cholinergic innervation from the basal forebrain, and GABAergic afferents from the VP/SI. Additionally, uncorrelated stochastic background input is applied to all model cells.
Input 1: thalamic/cortical glutamatergic afferentsWe modeled the thalamic and cortical glutamatergic afferents of the BLA as independent 2 Hz Poisson trains, which were delivered to the PNs, SOM + interneurons and CR + interneurons (Fig. 5A). Given the limited thalamic/cortical glutamatergic input received by PV + neurons (Smith et al. 2000) and the absence of any impact on model outcomes upon the removal of this connection, the schematic figures omit this ineffective connection.
Input 2: cholinergic innervationThe cholinergic innervation was simulated by changing the relevant synaptic conductance values, following prior work (Hummos et al. 2014). Three levels of cholinergic tone were modeled: low acetylcholine (ACh) (0), baseline ACh (1), and high ACh (2). For affected synapses, the synaptic current was multiplied by a factor as listed below, for both iampa and iGABA. For example, in the case of iampa, we get Eq. 6 below (replace ampa with GABA for the inhibitory synapses),
$$i_ = i_ *\left( *\left( \right)} \right).$$
(6)
Here, bACh and ACh together control the strength and sign for the various cholinergic conditions. For instance, an ACh value of 2 allows bACh to influence iampa positively, to make no change with ACh = 1, and to influence iampa negatively with ACh = −0.2. The specific bACh values (same for all ACh cases) and the corresponding synapses were as follows: 0.3 for all the background synapses (to PNs, and PV + , SOM + and CR + interneurons), 0.3 for PV + interneuron-PN and VP/SI-PN, 0.4 for SOM + interneuron-PN and CR + interneuron-PN, and 0.3 for VP/SI-PV + interneuron synapses.
Input 3: VP/SI GABAergic rhythmic innervationThe rhythmic GABAergic input from VP/SI was modeled by using previously described methods (Fink et al. 2015). The input was assigned a specific frequency, and each cell exhibited “jitter” in its response to the input, simulating intercellular variability. Jitter was Gaussian normal distributed (N) for each cell, with zero mean and SD \(_^\). The time of the jth event of neuron i was given by:
$$t_^ = jT + N\left( ^ } \right).$$
(7)
A total of 893 afferent cells were designed to individually exhibit independent 2 Hz Poisson activity. The afferents project onto 800 PN and 93 PV + interneurons with an average convergence of 1 and 10.1 cells, respectively (McDonald et al. 2011). Two states were considered for these afferents. In the baseline or non-modulated state, each afferent was independent at 2 Hz as above. For the theta-modulated state, the firing rate of the afferents were modulated with a sine wave:
$$r\left[ t \right] = A*(\sin \left( \right) + \phi ) + off ,$$
(8)
where \(A=\frac-1}\), \(f\) is the frequency, \(t\) is a vector representing time, \(\phi\) is the phase, \(off\) is the offset firing rate of the spike train being modulated and \(0<d<1\) is the depth of modulation which represents the amplitude of the sine wave relative to \(off\). We used a depth of modulation 0.7. To generate the spike train, a random vector x[t] was generated with values uniformly distributed between 0 and 1. A spike was generated if \(x\left[t\right]\le r\left[t\right]dt\) where dt in our case was 0.1.
For experiments with theta-modulated VP input, jitter was applied at 8 Hz. For all other experiments, no jitter was applied, and the VP input was 2 Hz Poisson.
Input 4: background input to all cellsIn order to replicate the membrane potential fluctuations observed in vivo, we implemented a point conductance input directly onto the soma, simulating stochastic background synaptic activity through the Ornstein–Uhlenbeck process (Destexhe et al. 2001). Specifically, stochastic background input had two independent components, excitatory and inhibitory, for PN and PV + , SOM + , and CR + cell groups, as modeled previously by our group (Feng et al.
留言 (0)