
記住我
Dear Editor,
Haim-Munk syndrome (HMS; OMIM #245010), also called keratosis palmoplantaris with periodontopathia and onychogryphosis or Cochin Jewish disorder, is an autosomal recessive rare disorder caused by a homozygous mutation in the CTSC gene encoding cathepsin C at chromosome11q14. It was first described in 1965 by Salim Haim, a dermatologist, and Munk, a radiologist among members of a small Jewish community from Kochi, India.1 This syndrome is characterised by palmoplantar keratoderma, early onset periodontitis, onychogryphosis, arachnodactyly, acro-osteolysis, and pes planus.2 Herein, we describe a case series of HMS in a single Indian consanguineous family, along with widespread ichthyosis, as an unusual association.
A 12-year-old girl, residing in Karnataka, South India, born of consanguineous parents [Figure S1], presented with palmoplantar keratoderma along with ichthyosis from 4 months of age. Antenatal, perinatal, and postnatal histories were non-contributory. There was no history of collodion membrane at birth. Her developmental milestones were normal. Her younger siblings, a 7-year-old girl, and a 4-year-old boy, had similar clinical symptoms from infancy. Additionally, similar complaints were reported in two maternal uncles. There was no systemic involvement in any child.
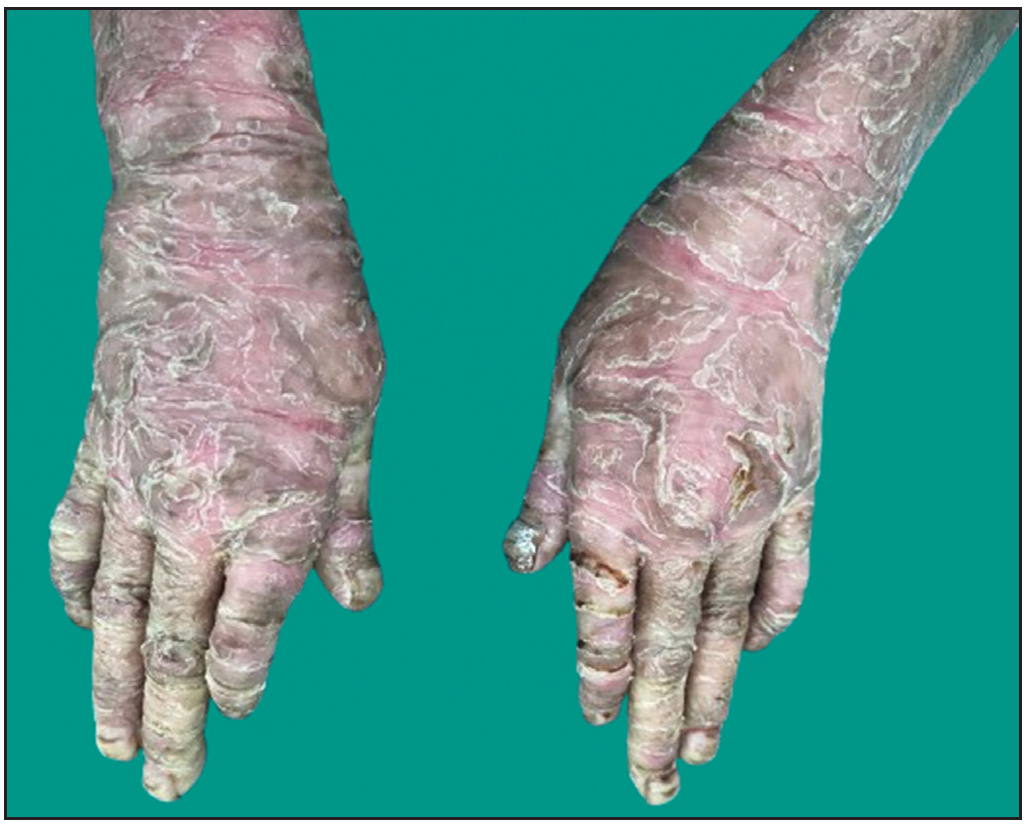
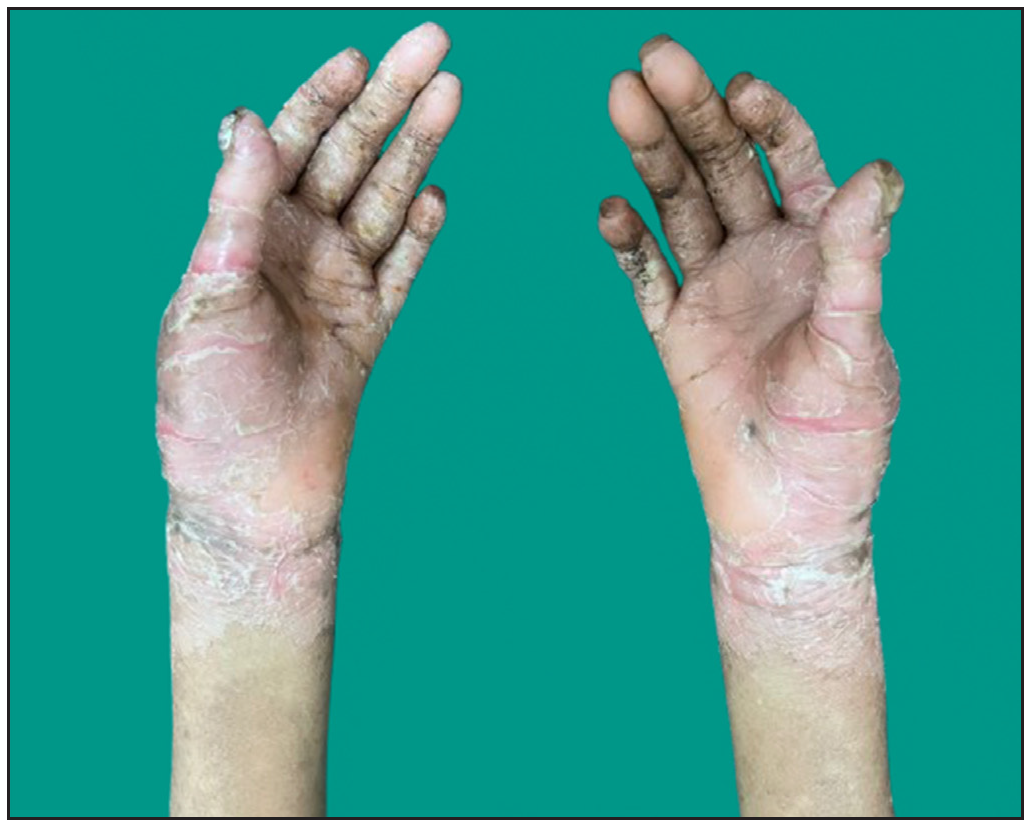
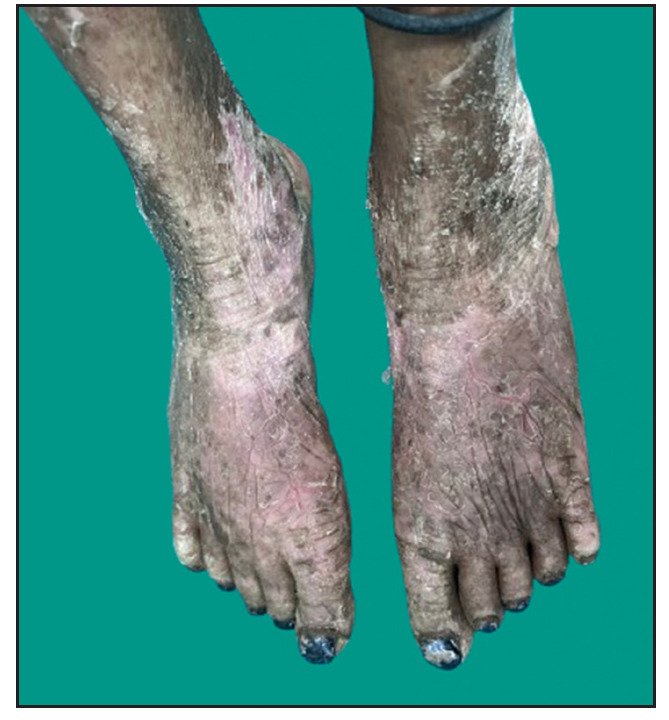
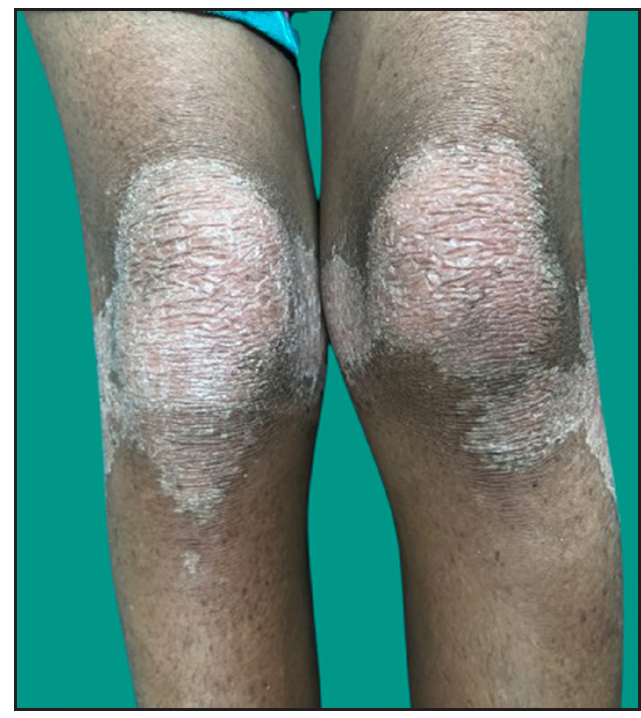
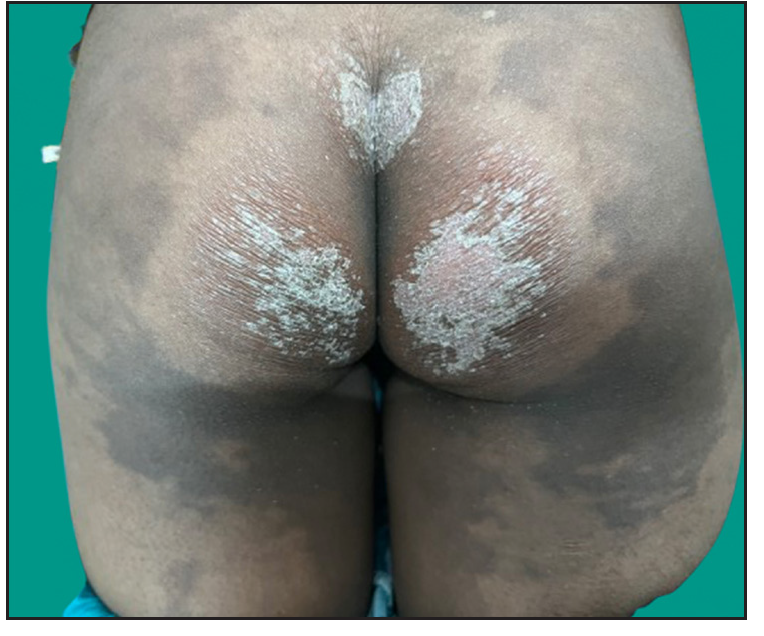
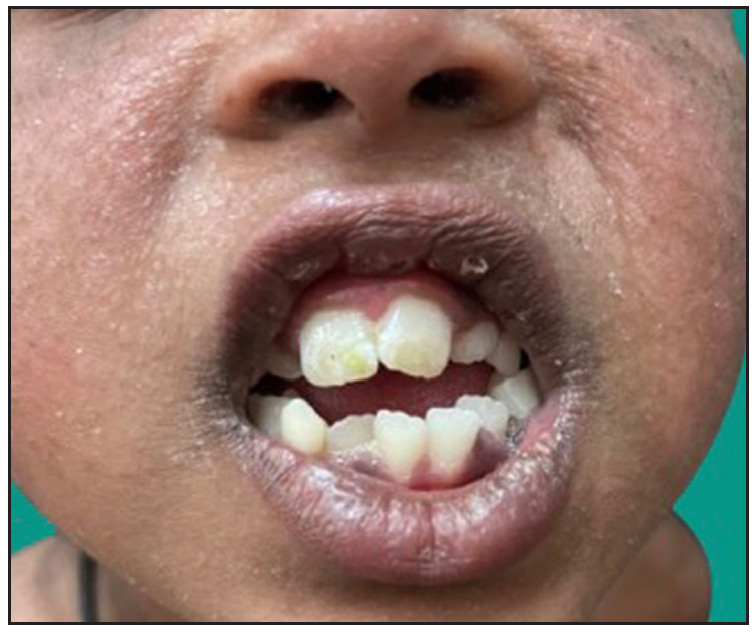
Dermatological examination of the proband showed thick erythematous plaques with deep fissures on the palmar and dorsal aspect of both hands and feet along with fixed contracture deformities involving all fingers [Figures 1a–c]. Well-defined psoriasiform plaques were present on the extensors of both elbows and knees and also on the gluteal region [Figures 1d–e]. In addition, other findings included thick scalp scaling, onychogryphosis of fingernails, and pes planus. Dental evaluation showed periodontitis, gingival hyperplasia and crowding of teeth. Her two younger siblings had similar cutaneous findings along with erythema and brown-plate-like scales all over the body [Figures 1f, S2, and S3].
Export to PPT
Export to PPT
Export to PPT
Export to PPT
Export to PPT
Export to PPT
Differential diagnoses of Papillon-Lefevre syndrome (PLS), loricrin keratoderma, and non-bullous ichthyosiform erythroderma were considered for all siblings. Complete haemogram, liver and renal function tests, fasting lipid and thyroid profile, serum electrolytes, serum calcium, and phosphorus were normal, except for insufficient vitamin D levels. Ultrasound abdomen and pelvis were normal in all subjects. A skeletal radiograph of both hands and feet of the proband showed arachnodactyly and pes planus [Figure S4]. Skeletal surveys of her younger siblings appeared normal, except for mild acro-osteolysis in the 7-year-old girl.
Genetic analysis done for the proband by next-generation sequencing technology detected a homozygous nonsense variant in exon 7 (c.1269G>A, p.Trp423Ter) of the CTSC gene in chromosome 11, resulting in a stop codon and premature truncation of the protein at codon 423, thus confirming the diagnosis of HMS [Figure S5]. The observed variant in our case causing HMS has not been reported in the 1000 genomes, gnomAD, topmed, and Clinvar databases and has been classified as a likely pathogenic variant based on the ACMG guidelines. We were unable to do the sequencing for the other two siblings and parents due to logistic constraints.
Cathepsin C (CTSC or dipeptidyl aminopeptidase I) is a lysosomal protease that can cleave dipeptides from the amino terminal of protein substrates. The CTSC gene is usually expressed in epithelial areas such as palms, soles, knees, and keratinised oral gingiva and also present in osteoclasts.3 Though PLS and HMS share similar genotype and phenotype, additional features like onychogryphosis, pes planus, arachnodactyly, and acro-osteolysis indicate HMS. Indeed, the cutaneous manifestations and the bone and finger deformities in HMS are worse and more extreme, whereas the periodontal involvement in HMS is less severe compared to PLS.4 PLS is a milder form, most commonly reported with missense mutations of the CTSC gene, whereas HMS is a severe phenotype, caused by nonsense mutations of the same gene. Thus, HMS may represent a variable phenotypic spectrum of CTSC mutations.5 Hitherto unknown genetic or environmental modifiers may be responsible for the phenotypic variability of this gene, making genotype and phenotype correlation difficult. In our case, though all the features were characteristic of PLS, presence of onychogryphosis, arachnodactyly, and pes planus favoured the diagnosis of HMS.
Fewer than 100 cases of HMS have been described in the literature, out of which genetic testing has been done in only 8 studies (n=19) [Table 1]. The variant described in our case has been reported before in PLS, as documented by Noack et al.5 However, to our knowledge, it has never before been linked to an HMS phenotype. Systemic involvement reported among the paediatric patients with genetically proven HMS includes liver abscess and arthritis in two studies by Hart et al.6 and McCarthy et al.7, respectively. None of our three siblings had systemic involvement.
Table 1: Genotype and phenotype correlation of all genetically confirmed cases of Haim-Munk syndrome
Study Year Numb er of patients Age/ sex Consan guinity PPK Periodontitis/premature shedding of teeth Nail (onychogr yphosis) Hands & feet Systemic involvement Family history Mutation analysis (CTSC gene) Hart et al.6 2000 11 - Present only in 1 out of 5 sibships + + Not mentioned AD One 15-year -old child with liver abscess + codon 286 of exon 6 (2127 A→G) Lidar et al.8 2004 1 35 yrs/F Not mention ed + + Not mentioned AD, AO Symmetric destructive arthritis of wrists and shoulders + codon 286 of exon 6 (2127 A→G) Cury et al.1 2005 1 19 yrs/F + + + + AD, AO, PP - - codon 196 of exon 4 (587T→C) Rai et al.3 2010 1 36 yrs/M + + + + AD, PP Liver abscess - exon 1 (145C→T Aswath et al.9 2014 1 23 yrs/M + -* + + - - - codon 453 of exon 7 (A→G) Sulak et al.2 2016 1 39 yrs/F Not mention ed + + + AD, PP - - exon 5 (748C→T) Wei et al.4 2020 2 14 yrs/M - + + + AD, AO - + exon 7 (1337A>C) 12 yrs/M - + + - - - + exon 7 (1337A>C) McCarthy et al.7 2023 1 14 yrs/M + + + + AD, PP Arthritis - (901G>A) Present case - 1 12 yrs/F + + + - AD, PP - + exon 7 (1269G>A)A multidisciplinary approach involving dermatologists, paediatricians, pedodontists, and physiotherapists is essential for the overall management of patients with HMS. Oral retinoid, acitretin at the dose of 0.5 mg/kg/day, along with topical emollients were prescribed for the elder two siblings along with referral to other departments, which resulted in improvement. The youngest sibling was given only topical emollients.
留言 (0)