
記住我
Short-rib thoracic dysplasia 7 (SRTD7) with or without polydactyly is a group of autosomal recessive skeletal ciliopathies with short ribs and thoracic stenosis as the main phenotypes. SRTD7 with or without polydactyly is caused by homozygous or compound heterozygous mutations in WDR35 gene. WDR35 (OMM 613602), a WD40 domain-containing protein encoding 28 exons, is located on chromosome 2q24.1, playing an important role in intraflagellar transport (1). It is typically characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a “trident” appearance of the acetabular roof, other organ malformations, such as renal cysts, which phenotypically overlap with cranioectodermal dysplasias (CED), an autosomal recessive disorder characterized by sagittal craniosynostosis, dolichocephaly, ectodermal abnormalities, skeletal dysplasia, characteristic facial features, and other clinical anomalies (2, 3).
WDR35 gene may also lead to CED. A previous study reported that SRTD7 affected the same organs as did CED with much greater severity, probably due to more severe mutations in WDR gene, such as deletion, non-sense mutations, or frameshift mutations (4). In this study, we report a 14.38-kb novel deletion compound combined with a causative variant in WDR35 gene leading to SRTD7 in a Chinese family. In addition, we also identified two compounded heterozygous variants in NPHP4 gene in the fetus of the said family, which may be partially responsible for fetal kidney hyperechogenicity and oligohydramnios.
Case presentationA 30-year-old G4P1 pregnant woman from Quanzhou, Southeast China, came to our hospital for genetic and etiological diagnosis at the gestational age of 18 weeks because of prenatal ultrasound anomalies. The couple denied consanguinity marriage and family-inherited diseases. Her first pregnancy was terminated because of short-limb deformities and congenital heart defects detected at the gestational age of 28 weeks. During her second pregnancy, similar ultrasound anomalies were observed and they chose to terminate the pregnancy. At her third pregnancy, no remarkable ultrasound anomalies were observed, and a full-term female infant was born who displayed normal developmental milestones at 5 years of age. This is her fourth pregnancy and several ultrasound anomalies were detected including short limbs, curved bones, narrow chest, enhanced renal echogenicity, oligohydramnios, enhanced intestinal echogenicity, and choroid plexus cyst (Figure 1). Finally, the family chose to terminate the pregnancy, and the aborted tissue was collected. After informed consent was signed, chromosomal microarray analysis (CMA) and whole exome sequencing (WES) were carried out to detect fetal copy number variants (CNVs) and sequence variants. No chromosomal abnormality was detected in the fetus by chromosomal microarray analysis.
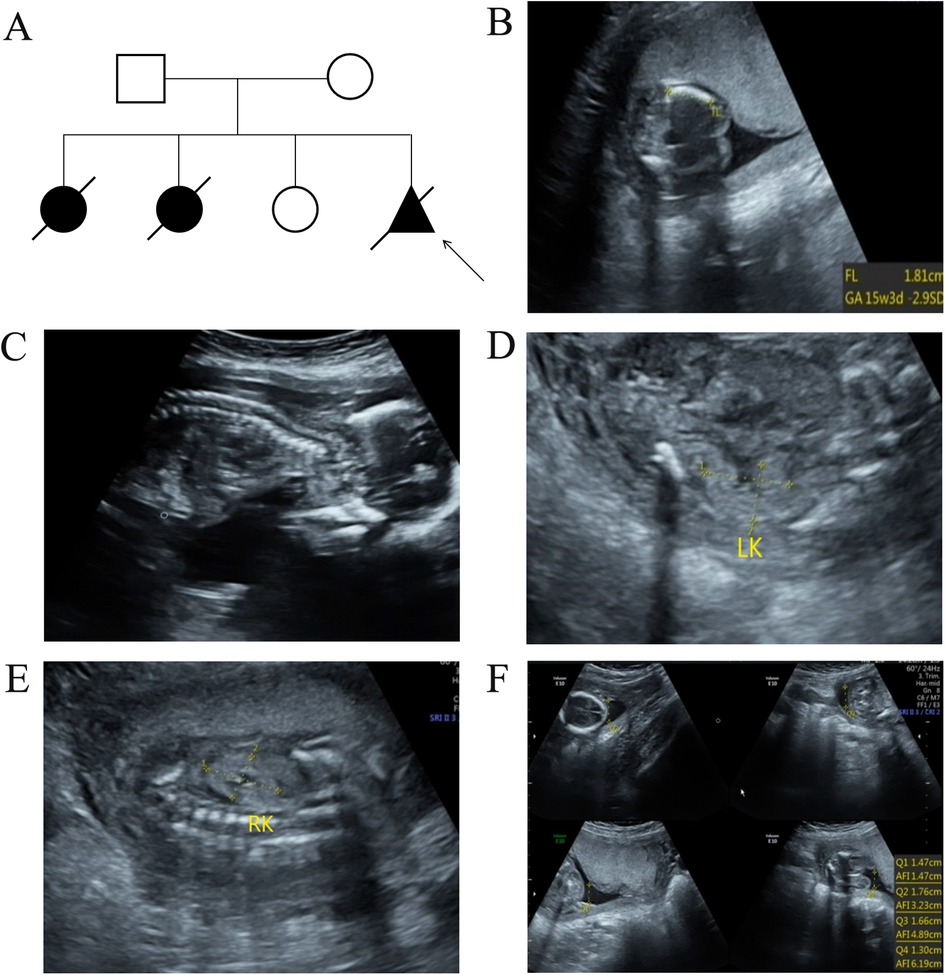
Figure 1. The pedigree information of the family and ultrasound examination results in the present fetus. (A) The pedigree analysis results of the enrolled family. The arrow indicates the proband. (B) Ultrasound examination results revealed short limbs and curved bones. (C) A narrow chest was also observed in the fetus. In addition, enhanced renal echogenicity (D,E) and oligohydramnios (F) was observed in the present fetus.
Further WES was carried out to investigate relevant sequence variants in the affected fetus. The result revealed a 14.38-kb novel deletion compound covering exon 7 to exon 12, which was combined with a missense variant NM_001006657.2:c.932G>T(p.W311l) in WDR35 in the fetus, which was believed to have been transmitted from the parent. According to the American College of Medical Genetics and Genomics (ACMG) guidelines, both variants were supposed to be pathogenic, which was further confirmed by Sanger sequencing and real time quantitative PCR (qPCR) (Figure 2). In addition, two compound heterozygous variants NM_015102.5:c.[1196A>G(p.E399G)];[1972C>T(p.R658*)] in NPHP4 gene were also identified in the fetus, which were inherited from the parents, respectively. Both variants were further verified by Sanger sequencing (Figure 3). According to the ACMG guidelines, the variant of NM_015102.5:c.1972C>T(p.R658*) was classified as a pathogenic variant (PVS1 + PP1_Strong + PM2_Supporting + PM3_Supporting), and c.1196A>G(p.E399G) was interpreted as a variant of unknown significance. As predicted by Spidex software, the variant of c.1196A>G(p.E399G) may affect splicing (PP3).
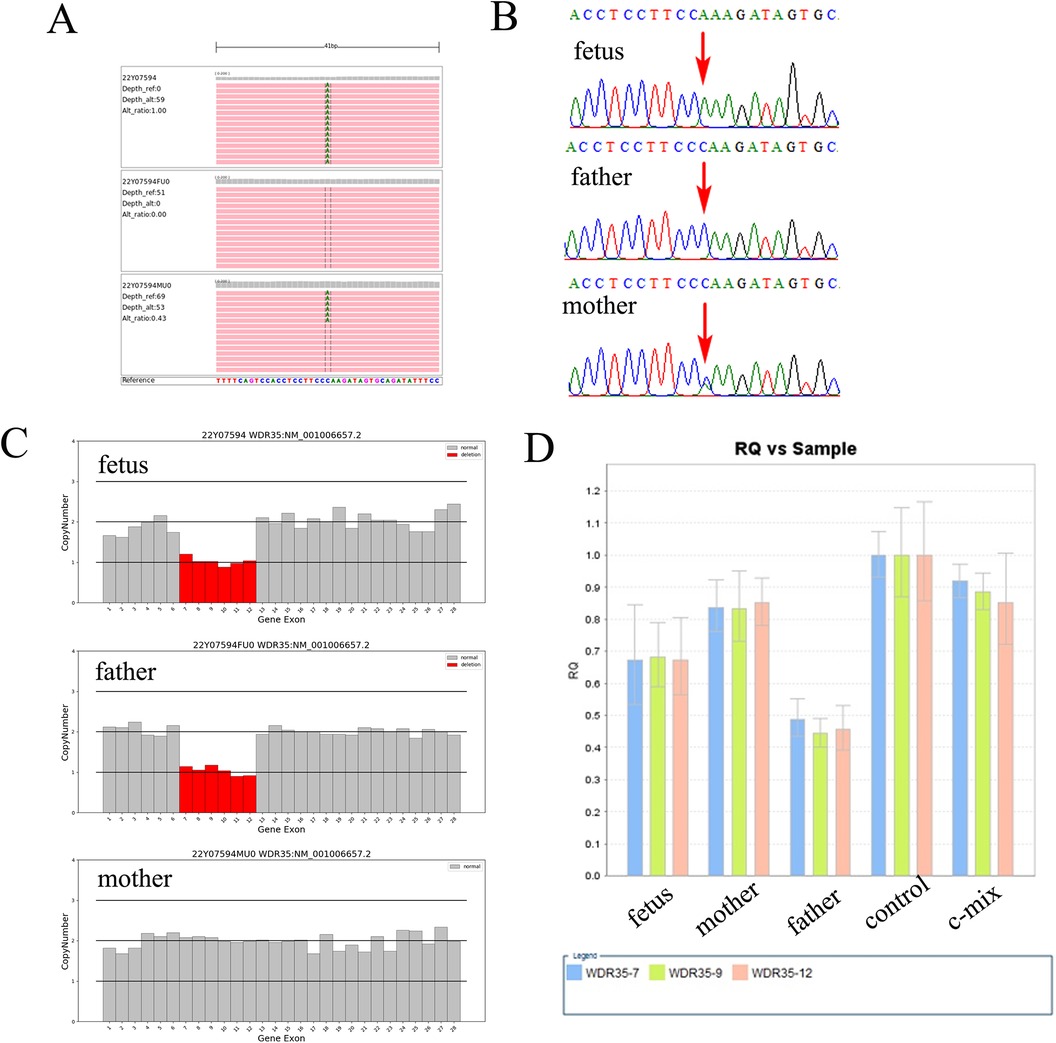
Figure 2. A 14.38-kb novel deletion compound with a missense variant in WDR35 is identified in the fetus. (A) Trio-whole exome sequencing (trio-WES) demonstrated a missense variant NM_001006657.2:c.932G>T(p.W311l) in WDR35 in the fetus. (B) Sanger sequencing verified the c.932G>T in the fetus, which was also present in the mother. (C) A 14.38-kb deletion compound encompassing exon 7 to exon 12 was also identified in the fetus using trio-WES. (D) qPCR analysis revealed a heterozygous deletion from exon 7 to exon 12 in WDR35 in both the fetus and the father, but the fetus's mother did not have the deletion.
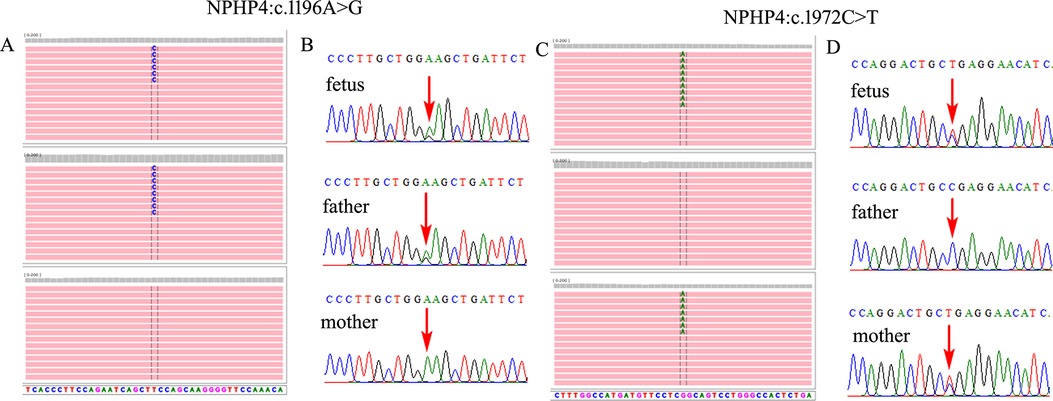
Figure 3. Two compounded heterozygous variants in NPHP4 gene were identified in the fetus. (A,C) Trio-WES demonstrated an NM_015102.5:c.1196A>G(p.E399G) compounded with NM_015102.5:c.1972C>T(p.R658*) in NPHP4 gene in the fetus, which was inherited from the parents, respectively. (B,D) Sanger sequencing verified both of the variants in the family.
Discussion and conclusionWES has become an increasingly popular technique for prenatal etiological diagnosis of genetic causes of fetal structural abnormalities including multiple fetal malformations, skeletal dysplasia, central nervous system (CNS) abnormalities, cardiovascular malformations, and other structural anomalies (5, 6). Previous research has shown that WES can offer a high positive detection rate for skeletal dysplasia (7, 8). In addition, a most recent study conducted by Zeng et al. (9) indicated that clinical exome sequencing exhibits an obvious advantage in detecting small CNVs over CMA. Skeletal ciliopathy is a rare disease characterized by abnormalities in the skeletal system, caused by genetic defects that regulate the structure or function of primary cilia. In the present study, we identified a large novel deletion compound with a causative variant in WDR35 gene causing SRTD7 in a fetus with skeletal dysplasia, and two compound heterozygous variants in NPHP4 gene, which may be partially responsible for fetal kidney hyperechogenicity and oligohydramnios.
High genetic heterogeneity is present in SRTD. SRTD7 was first described by Kannu et al. (10) in a New Zealand family classified as an “unclassifiable” short-rib polydactyly syndrome. Caparrós-Martín et al. (11) studied five patients from three unrelated families with short ribs, mesomelic shortening of limbs, and tooth and nail dysplasia, and homozygosity splice mutations in WDR35 gene were identified. In addition, a previous study conducted by Duran et al. (12) reported three siblings and an unrelated female infant with compound heterozygosity mutations in WDR35 gene leading to SRTD7. All three sibs exhibited short ribs, short limbs, bilateral postaxial polydactyly of the hands and feet with aphalangia of the hands, and bending of humeri, radii, and ulnae. However, the unrelated female infant did not have polydactyly. The fetus in the present study was found to have short limbs, curved bones, and narrow chest on prenatal ultrasound imaging, all of which were consistent with the clinical features of SRTD7, but no polydactyly was detected in this fetus. CED, which is also caused by WDR35 mutations but with milder phenotypes, is mainly characterized by craniosynostosis and ectodermal abnormalities. In addition, most WDR35 gene variants leading to CED are missense mutations (4, 13, 14). By contrast, at least one allele in WDR35 gene had a loss-of-function in patients with SRTD7 (11, 12, 15). In the present study, a previous reported causative missense variant (c.932G>T) (16) compound with a large novel deletion in WDR35 gene was identified, which provides more evidence the cause of SRTD7. In addition, this family also experienced two pregnancies with fetal short limbs, which may be due to compound heterozygous mutations in WDR35 gene, but there is a lack of further genetic analysis.
In the present study, we also identified two compound heterozygous variants NM_015102.5:c.[1196A>G(p.E399G)];[1972C>T(p.R658*)] in NPHP4 gene in the fetus. Nephronophthisis is an autosomal recessive kidney disease characterized by the multicystic dysplastic kidney, oligohydramnios, and tubulointerstitial nephritis that progresses to end-stage renal disease (17). At present, four genes (NPHP1, NPHP2, NPHP3, and NPHP4) have been identified as being responsible for nephronophthisis. NPHP4 gene encoding nephrocystin-4 is known to cause end-stage renal disease in children and young adults (18). The pathogenic variant of c.1972C>T(p.R658*) has been identified in patients with nephronophthisis 4 and as being co-segregated with the disease in the family (19, 20). However, the c.1196A>G(p.E399G) variant was interpreted as an unclassified variant, but we cannot rule out that the detected NPHP4 variants may partially be responsible for fetal kidney hyperechogenicity and oligohydramnios.
The variants identified in WDR35 and NPHP4 of the fetus in this study may be responsible for the ultrasound anomalies identified in the second trimester of pregnancy. However, further functional analysis is required to explore the pathogenicity of the c.1196A>G(p.E399G) variant in NPHP4. In addition, the specimen and specific clinical phenotypes of the previous two fetuses in this pregnant woman that had similar clinical abnormalities were not available in this study.
In this study, we presented a novel heterozygous compound variant in WDR35 gene causing SRTD7 in a Chinese family, which may provide more insights into understanding the genotype and phenotype correlation. In addition, we also identified two compounded heterozygous variants in NPHP4 gene in the fetus, which may be partially responsible for fetal kidney hyperechogenicity and oligohydramnios.
Data availability statementThe raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statementEthical approval was obtained from the Institutional Ethics Committee of Quanzhou Women's and Children's Hospital for commencement of this study involving humans (2020 No. 31). The study was conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributionsJZ: Funding acquisition, Investigation, Writing – original draft, Writing – review & editing. JW: Investigation, Writing – original draft. ZH: Writing – original draft. YC: Formal Analysis, Investigation, Writing – original draft. CC: Writing – review & editing, Formal Analysis, Methodology.
FundingThe author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was sponsored by Huaqiao University Joint of Hospital and University Innovation Project (2023YX001) and Quanzhou City Science & Technology Program of China (2023NS068).
AcknowledgmentsWe express our appreciation for the patient and other subjects who participated in this study. We also wish to express our appreciation for the Huaqiao University and Quanzhou City Science and Technology Bureau for funding this work.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statementThe authors declare that no Generative AI was used in the creation of this manuscript.
Publisher's noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References1. Gilissen C, Arts HH, Hoischen A, Spruijt L, Mans DA, Arts P, et al. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am J Hum Genet. (2010) 87(3):418–23. doi: 10.1016/j.ajhg.2010.08.004
PubMed Abstract | Crossref Full Text | Google Scholar
2. Ryzko J, Walczak-Sztulpa J, Czubkowski P, Latos-Bielenska A, Kowalski A, Stefanowicz M, et al. Case report: sequential liver after kidney transplantation in a patient with Sensenbrenner syndrome (cranioectodermal dysplasia). Front Pediatr. (2022) 10:834064. doi: 10.3389/fped.2022.834064
PubMed Abstract | Crossref Full Text | Google Scholar
3. Antony D, Nampoory N, Bacchelli C, Melhem M, Wu K, James CT, et al. Exome sequencing for the differential diagnosis of ciliary chondrodysplasias: example of a WDR35 mutation case and review of the literature. Eur J Med Genet. (2017) 60(12):658–66. doi: 10.1016/j.ejmg.2017.08.019
PubMed Abstract | Crossref Full Text | Google Scholar
4. Hoffer JL, Fryssira H, Konstantinidou AE, Ropers HH, Tzschach A. Novel WDR35 mutations in patients with cranioectodermal dysplasia (Sensenbrenner syndrome). Clin Genet. (2013) 83(1):92–5. doi: 10.1111/j.1399-0004.2012.01880.x
PubMed Abstract | Crossref Full Text | Google Scholar
5. Monaghan KG, Leach NT, Pekarek D, Prasad P, Rose NC, ACMG Professional Practice and Guidelines Committee. The use of fetal exome sequencing in prenatal diagnosis: a point to consider document of the American College of Medical Genetics and Genomics (ACMG). Genet Med. (2020) 22(4):675–80. doi: 10.1038/s41436-019-0731-7
PubMed Abstract | Crossref Full Text | Google Scholar
6. Application Collaboration Group of Whole Exome Sequencing in Prenatal Diagnosis, Lou G, Hou Q, Yang K, Guo L. Expert consensus on the application of prenatal exome sequencing for fetal structural anomalies. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. (2022) 39(5):457–63. doi: 10.3760/cma.j.cn511374-20210920-00765
PubMed Abstract | Crossref Full Text | Google Scholar
7. Lord J, McMullan DJ, Eberhardt RY, Rinck G, Hamilton SJ, Quinlan-Jones E, et al. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet. (2019) 393(10173):747–57. doi: 10.1016/S0140-6736(18)31940-8
PubMed Abstract | Crossref Full Text | Google Scholar
8. Fu F, Li R, Yu Q, Wang D, Deng Q, Li L, et al. Application of exome sequencing for prenatal diagnosis of fetal structural anomalies: clinical experience and lessons learned from a cohort of 1,618 fetuses. Genome Med. (2022) 14(1):123. doi: 10.1186/s13073-022-01130-x
PubMed Abstract | Crossref Full Text | Google Scholar
9. Zeng Y, Ding H, Wang X, Huang Y, Liu L, Du L, et al. High positive predictive value of CNVs detected by clinical exome sequencing in suspected genetic diseases. J Transl Med. (2024) 22(1):644. doi: 10.1186/s12967-024-05468-1
PubMed Abstract | Crossref Full Text | Google Scholar
10. Kannu P, McFarlane JH, Savarirayan R, Aftimos S. An unclassifiable short rib-polydactyly syndrome with acromesomelic hypomineralization and campomelia in siblings. Am J Med Genet A. (2007) 143A(21):2607–11. doi: 10.1002/ajmg.a.31989
PubMed Abstract | Crossref Full Text | Google Scholar
11. Caparrós-Martín JA, De Luca A, Cartault F, Aglan M, Temtamy S, Otaify GA, et al. Specific variants in WDR35 cause a distinctive form of Ellis-Van Creveld syndrome by disrupting the recruitment of the EvC complex and SMO into the cilium. Hum Mol Genet. (2015) 24(14):4126–37. doi: 10.1093/hmg/ddv152
PubMed Abstract | Crossref Full Text | Google Scholar
12. Duran I, Taylor SP, Zhang W, Martin J, Qureshi F, Jacques SM, et al. Mutations in IFT-A satellite core component genes IFT43 and IFT121 produce short rib polydactyly syndrome with distinctive campomelia. Cilia. (2017) 6:7. doi: 10.1186/s13630-017-0051-y
PubMed Abstract | Crossref Full Text | Google Scholar
13. Bacino CA, Dhar SU, Brunetti-Pierri N, Lee B, Bonnen PE. WDR35 mutation in siblings with Sensenbrenner syndrome: a ciliopathy with variable phenotype. Am J Med Genet A. (2012) 158A(11):2917–24. doi: 10.1002/ajmg.a.35608
PubMed Abstract | Crossref Full Text | Google Scholar
14. Smith C, Lamont RE, Wade A, Bernier FP, Parboosingh JS, Innes AM. A relatively mild skeletal ciliopathy phenotype consistent with cranioectodermal dysplasia is associated with a homozygous nonsynonymous mutation in WDR35. Am J Med Genet A. (2016) 170(3):760–5. doi: 10.1002/ajmg.a.37514
PubMed Abstract | Crossref Full Text | Google Scholar
15. Mill P, Lockhart PJ, Fitzpatrick E, Mountford HS, Hall EA, Reijns MA, et al. Human and mouse mutations in WDR35 cause short-rib polydactyly syndromes due to abnormal ciliogenesis. Am J Hum Genet. (2011) 88(4):508–15. doi: 10.1016/j.ajhg.2011.03.015
PubMed Abstract | Crossref Full Text | Google Scholar
16. Toriyama M, Lee C, Taylor SP, Duran I, Cohn DH, Bruel AL, et al. The ciliopathy-associated CPLANE proteins direct basal body recruitment of intraflagellar transport machinery. Nat Genet. (2016) 48(8):970. doi: 10.1038/ng0816-970b Nat Genet. (2016) 48(6):648–656.27463398
PubMed Abstract | Crossref Full Text | Google Scholar
17. Ijaz A, Alfadhli F, Alharbi A, Khan YN, Alhawas YK, Hashmi JA, et al. NPHP3 splice acceptor site variant is associated with infantile nephronophthisis and asphyxiating thoracic dystrophy; A rare combination. Eur J Med Genet. (2022) 65(10):104578. doi: 10.1016/j.ejmg.2022.104578
PubMed Abstract | Crossref Full Text | Google Scholar
18. Otaki Y, Watanabe T, Sato J, Kobayashi Y, Aono T, Saito Y, et al. Association of nephronophthisis 4 genetic variation with cardiorenal syndrome and cardiovascular events in Japanese general population: the Yamagata (Takahata) study. Heart Vessels. (2022) 37(4):673–82. doi: 10.1007/s00380-021-01953-5
PubMed Abstract | Crossref Full Text | Google Scholar
19. Otto E, Hoefele J, Ruf R, Mueller AM, Hiller KS, Wolf MT, et al. A gene mutated in nephronophthisis and retinitis pigmentosa encodes a novel protein, nephroretinin, conserved in evolution. Am J Hum Genet. (2002) 71(5):1161–7. doi: 10.1086/344395
PubMed Abstract | Crossref Full Text | Google Scholar
20. Chaki M, Hoefele J, Allen SJ, Ramaswami G, Janssen S, Bergmann C, et al. Genotype-phenotype correlation in 440 patients with NPHP-related ciliopathies. Kidney Int. (2011) 80(11):1239–45. doi: 10.1038/ki.2011.284
留言 (0)