
記住我
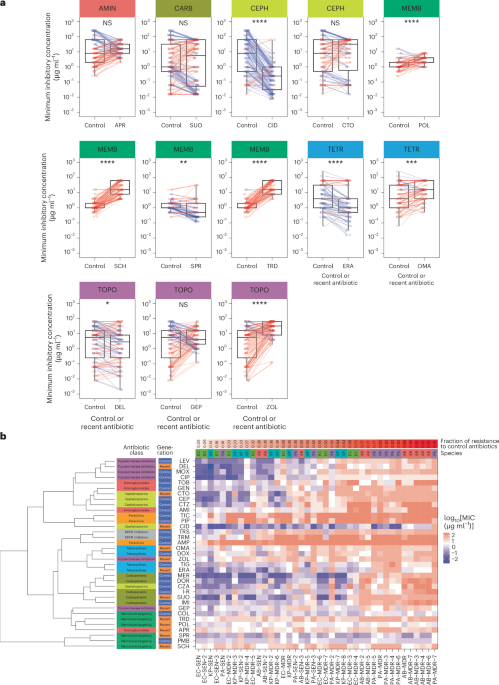




This study focused on multiple bacterial strains. We tested the activity spectrum of the antibiotics in this study on a set of 40 clinically relevant pathogenic strains of 4 species (E. coli, K. pneumoniae, A. baumannii and P. aeruginosa; see the whole list of pathogens in Supplementary Table 4). For the FoR and ALE experiments, two strains per species were chosen: for SEN strains, E. coli ATCC 25922, K. pneumoniae ATCC 10031, A. baumannii ATCC 17978 and P. aeruginosa ATCC BAA-3107; and for MDR strains, E. coli NCTC 13846, K. pneumoniae ATCC 700603, A. baumannii ATCC BAA-1605 and P. aeruginosa LESB58. For E. coli, we chose the ATCC 25922 strain as a SEN strain due to its widespread use in the literature, and an mcr1-carrying NCTC 13846 strain as the MDR strain due to the high interest in the impact of this mobile resistance gene on colistin resistance43. For the other three species, SEN and MDR strains were selected based on the highest number of control antibiotics to which they showed sensitivity or resistance, respectively, with the additional criterion for MDR strains that they should be part of an official strain collection (Extended Data Fig. 3). Functional metagenomic screens were performed with E. coli ATCC 25922 and K. pneumoniae ATCC 10031 strains. Deep-scanning mutagenesis (DIvERGE) was performed with E. coli K12 MG1655 and K. pneumoniae ATCC 10031 strains. The collection of clinical samples was performed in a previous study44. No new samples were collected for the current study. The collection of clinical samples complies with all relevant ethical regulations and was approved by the Scientific and Research Ethics Committee of the Hungarian Health Science Council (BMEÜ /271-3/2022/EKU). As specified in Material Transfer Agreements, these isolates and their derivatives cannot be transferred to a third party as they can be used only at the recipient organizations and only in the recipient scientists’ laboratories under the direction of the recipient scientists or others working under their direct supervision.
A total of 19 antibiotics were applied in this study from 6 different antibiotic families: 13 newly developed (recent) antibiotics, which are in different phases of clinical trials, and 6 conventional (control) antibiotics with a long clinical history from each antibiotic family. For names, abbreviations and further details, see Table 1 and Supplementary Table 3. Antibiotics were custom-synthesized or purchased from several distributors (Supplementary Table 3). For preparation, each antibiotic stock solution was filter-sterilized and kept at −20 °C until use. For more details on recent and control antibiotics, see Supplementary Note 1, Table 1 and Supplementary Table 3. For data on clinical breakpoints and peak plasma concentrations, see Supplementary Table 3. Tridecaptin M152-P3 was synthesized using standard Fmoc-based solid-phase peptide synthesis. The process began with resin preparation, followed by iterative Fmoc deprotection and amino acid coupling steps using HBTU as the coupling reagent. After assembly of the peptide chain, the product was cleaved from the resin using a TFA-based cocktail, precipitated with cold isopropyl ether, and dried under vacuum. The crude peptide was then purified using preparative HPLC, yielding 233.4 mg of the product with 98.17% purity as confirmed by LC-MS and HPLC.
The synthesis of POL7306 involved three main stages: the preparation of two intermediate peptides (Compound 1 and Compound 2) and their subsequent connection. Fmoc-based solid-phase peptide synthesis was used, beginning with the attachment of amino acids to a resin, followed by deprotection and coupling steps. After peptide assembly, the crude peptides were cleaved from the resin and purified by precipitation and washing with isopropyl ether. The final connection of the two peptides was performed on a Rink Amide resin, followed by cleavage with TFA and purification through prep-HPLC. The final product, POL7306, was obtained with 96.71% purity and confirmed by LCMS and HPLC.
For detailed synthesis route see Supplementary information.
Unless otherwise indicated, cation-adjusted Mueller–Hinton broth 2 (MHB; Millipore) medium was used throughout the study, except for cefiderocol and the folate biosynthesis inhibitor SCH-79797. Following the European Committee on Antimicrobial Susceptibility Testing’s (EUCAST) recommendation on cefiderocol, iron-depleted MHB media was used45. To maximize antibacterial activity of SCH-79797, based on previous experience with folate biosynthesis inhibitor antibiotics46, Minimal Salt (MS) medium was used (1 g l−1 (NH4)2SO4, 3 g l−1 KH2PO4 and 7 g l−1 K2HPO4 supplemented with 1.2 mM Na3C6H5O7·2H2O, 0.4 mM MgSO4, 0.54 μg ml−1 FeCl3, 1 μg ml−1 thiamine hydrochloride, 0.2% casamino acids and 0.2% glucose).
High-throughput MIC measurementsA standard serial broth microdilution technique47 was used to determine MICs, as suggested by the Clinical and Laboratory Standards Institute guidelines. A robotic liquid-handling system was used to automatically prepare 11–16 step serial dilutions in 384 well microtiter plates. A total of 5 × 105 bacterial cells per ml were inoculated into each well containing 60 µl medium. Bacterial cultures were incubated at 37 °C with continuous shaking (300 rpm) for 18 h (2 replicates from each). Cell growth was monitored by measuring the optical density (optical density at 600 nm (OD600) values using a Biotek Synergy microplate reader). MIC was defined as the antibiotic concentration of complete growth inhibition (that is, OD600 < 0.05). The same protocol was used to estimate antibiotic susceptibility of laboratory-evolved lineages. Relative MIC was calculated as follows: log2(MICevolved/MICancestor). Increase in MIC was calculated as follows: log10(MICevolved − MICancestor).
We aimed to perform both FoR and ALE assays with all selected 8 bacterial strains; however, 34% of all antibiotic–strain combinations (N = 52) were excluded from further experiments due to modest initial drug efficacy (that is, MIC > 4 µg ml−1), rendering them less relevant for clinical use. The prevalence of panel strains with reduced susceptibility to a certain antibiotic was estimated by calculating the fraction of panel strains with high initial MIC values.
FoR assaysTo estimate the frequency of spontaneous mutations that confer resistance in a microbial population, the FoR assay was used. Using standard protocols16,17,18,19, approximately 1010 cells from stationary-phase cultures were plated to antibiotic-containing MHB plates. Before plating, bacteria were grown overnight in MHB medium at 37 °C, 250 rpm, collected by centrifugation (3,880g for 10 min) and washed once in equal volumes of MHB. From this concentrated bacteria suspension, ~1010 cells were plated to agar plates containing the selective drug at the desired concentration (that is, 2×, 4×, 8× and 20× MIC of each given antibiotic). Unless otherwise indicated (see ‘High-throughput MIC measurements’ above), MHB agar medium was used throughout the study. All experiments were performed in three replicates. Plates were grown at 37 °C for 48 h. Total colony-forming units were determined simultaneously in each experiment by plating appropriate dilutions to antibiotic-free MHB agar plates. Resistance frequencies for each bacterial strain were calculated by dividing the number of emergent colonies by the initial viable cell count. Ten bacterial colonies from the highest antibiotic concentration were selected for further MIC measurements and whole-genome sequence analysis.
High-throughput ALEA previously established protocol48,49 was used for ALE, with the aim to ensure that populations with the highest level of resistance were propagated further. Starting with an antibiotic concentration resulting in ~50% growth inhibition, 10 parallel populations per antibiotic–ancestor strain combination were grown for 72 h at 37 °C with continuous shaking (300 rpm). As rapid degradation has been observed for β-lactams and cephalosporins in liquid laboratory media50, ceftobiprole, cefiderocol and cefepime were not subjected to ALE. Unless otherwise indicated, MHB medium was used. After each incubation period, 20 μl of each bacterial culture was transferred to 4 new independent wells containing freshly prepared medium containing different antibiotic concentrations (0.5×, 1×, 1.5× and 2.5× the concentration of the previous step). A chessboard layout was used on the plate to monitor potential cross-contamination events. Cell growth was monitored before each transfer by measuring the OD600 value (Biotek Synergy 2 microplate reader). Only populations with the highest drug concentration (and reaching OD600 > 0.2) were selected for further transfer. The evolution experiment was generally continued for 20 transfers, resulting in a total of 728 evolved lines (78 lines were omitted because of limited growth).
Whole-genome sequencingTo identify potential antibiotic-resistance-conferring mutations, we generally selected two to five lines from FoR and ALE experiments, respectively, for whole-genome sequencing. Resistant populations were grown overnight in antibiotic-free medium. DNA isolation from overnight cultures was performed with the GenElute Bacterial Genomic DNA Kit (Sigma), according to the manufacturer’s instructions. DNA was eluted with 120 µl RNAse-free sterile water in 2 elution steps. The eluted DNA (60 µl) was then concentrated using the DNA Clean and Concentrator Kit (Zymo), according to the manufacturer’s instructions. The final DNA concentration was measured using a Qubit Fluorometer and concentration was set to 1 ng ml−1 in each sample. Sequencing libraries from isolated genomic DNA were prepared using the Nextera XT DNA Library Preparation Kit (Illumina) following the manufacturer’s instructions. The sequencing libraries were sequenced on an Illumina NextSeq 500 sequencer using mid or high output flow cells to generate 2× 150 bp paired-end reads.
To determine and annotate the variants, we mapped the sequencing reads to their corresponding reference genomes using an established method (Burrows–Wheeler Aligner)51. From the aligned reads, PCR duplicates were removed with the Picard MarkDuplicates tool (http://broadinstitute.github.io/picard/). We removed every read that had been aligned with more than six mismatches (disregarding insertions and deletions). The SNPs and insertions or deletions were called using Freebayes52 with the following parameters: -p 5–min-base-quality 28. The identified variants were filtered using the vcffilter tool from vcflib53 using the following parameters: –f ‘QUAL > 100’. To avoid missing rare but valid hits, we did not set a lower limit for the prevalence of rare variants. If necessary, mutations were also manually inspected within the aligned reads using IGV54 to reduce Burrows–Wheeler alignment or freebayes artefacts. Finally, the variants were annotated with SnpEf and we only kept those that were not present in the ancestor. We filtered out mutations that appeared in more than nine lines because these variants are probably already present in the ancestor. Furthermore, mutations that appeared in less than nine but more than six lines were manually inspected to exclude sequencing artefacts. We also excluded mutations that affect 40 bp or longer repetitive regions, resulting in a filtered set of mutations. Lines containing mutations in the mutL, mutS or mutY genes, or lines with more than 19 mutations, defined by outlier filtering (third quartile + (3 × interquartile range)), were considered hypermutators and were subsequently discarded.
To analyse the presence or absence of mutations across genes and strain backgrounds, we first complemented the existing gene functional annotation for the eight bacterial strain backgrounds as follows. Nucleotide sequence and annotation files of six strains (E. coli ATCC 25922, K. pneumoniae ATCC 10031, A. baumannii ATCC 17978, P. aeruginosa ATCC BAA-3107, K. pneumoniae ATCC 700603 and A. baumannii ATCC BAA-1605) were downloaded from the ATCC database (https://www.atcc.org/). For P. aeruginosa LESB58 and E. coli NCTC 13846 strains, genomic data were downloaded from NCBI Nucleotide (accession numbers FM209186.1 and GCA_900448335.1). Next, all genes in the GeneBank files, including hypothetical ones, were functionally annotated using PANNZER2 (refs. 55,56). To compare the sets of mutated genes across strain backgrounds, we determined genes that are shared across different strains by identifying groups of orthologous genes using OrthoFinder (v.2.5.4)57,58.
Bioinformatic analysis of mutations promoting growth in the laboratory based on previous workWe compiled a comprehensive list of 104 genes associated with medium adaptation in E. coli, as identified in 2 previous studies59,60. First, we examined our evolved E. coli strains for mutations within these genes. The DNA sequences of these genes from the E. coli strain MG1655 were retrieved from EcoCyc (v.26.0)61. We then aligned the sequences to the amino acid sequences of proteins in our reference genomes (SEN and MDR, separately) using the BLASTX tool (implemented in the rBLAST R package62). For each gene, we selected the alignment with the highest bit score, requiring a sequence identity of at least 80% and coverage of at least 80%. This approach resulted in 92 of the 104 genes being matched in each reference genome. Among these, 8 genes in the MDR strains showed 13 mutations and 7 genes in the SEN strains contained 15 mutations, totalling 11 genes with 28 mutations in at least 1 strain. As a next step, we investigated non-coding mutations in our evolved strains to ascertain if any were located in or adjacent to operons overlapping with the genes implicated in medium adaptation. This analysis did not reveal any non-coding mutations associated with the genes of interest.
Comparison of variants to public genomes of bacterial isolatesWe assessed whether amino acid substitutions occurring in the FoR and ALE samples are present in natural populations of E. coli and A. baumannii as follows. We compiled a comprehensive genomic dataset for E. coli strains by downloading assembled genome sequences or unassembled reads and metadata, from four sources: (1) the JGI Integrated Microbial Genomes and Microbiomes (IMG) database63 (on 29 January 2020), (2) the NCBI Prokaryotic RefSeq collection (available at https://www.ncbi.nlm.nih.gov/refseq/; on 29 January 2020), (3) genomes that were analysed in ref. 64, and (4) genomes that were analysed in ref. 65.
After trimming the adaptors with the Cutadapt v.3.2 program66, we de novo assembled the next-generation sequencing short reads (downloaded from the Sequence Read Archive database67) of genomes from sources (3) and (4) using the SPAdes v.3.14.1 software68. Then we applied the BUSCO v.5.0.0. workflow69 to exclude genome sequences with less than 95% of the BUSCO genes, indicating inadequate completeness or quality. When multiple genome sequences belonged to the same BioSample identifier, only the one with the highest BUSCO score, longest sequence and fewest contigs were kept, and all metadata of the original sequences were merged. This resulted in 20,786 E. coli genomes (Supplementary Data 1) for which gene prediction was performed using Prodigal (v.2.6.3)70 to obtain protein-coding gene annotations that are consistent across the genomes. ORFs with less than 100 amino acids were filtered out. Strains were classified as pathogens and non-pathogens based on their genomic metadata. For A. baumannii strains, we downloaded all available assembled genome sequences from the NCBI Prokaryotic RefSeq genome collection (https://www.ncbi.nlm.nih.gov/refseq/) on 12 September 2022. Then we applied genome filtering with 95% completeness using BUSCO v.5.4.6. and protein prediction using Prodigal as described above for E. coli. This resulted in 15,185 A. baumannii genomes (Supplementary Data 1).
Next, we searched for the presence of each amino-acid-changing SNP across the E. coli and Acinetobacter genome collections as follows. First, we performed a sequence similarity search of each gene carrying a given variant using DIAMOND BLAST (v.2.0.2)71 using an e-value (expect-value) of 0.00001 with 90% coverage and 90% identity to identify homologues among the genomes. In the next step, we performed multiple sequence alignment using MAFFT (v.7.475)72 with the –retree 2 option. Then we analysed the amino acid frequency across the alignments in all mutated positions. All E. coli and A. baumannii variants that were present in the corresponding species’ genome collection and appeared more than once in our FoR and ALE samples were selected for further analysis.
Functional metagenomic screensResistance-conferring DNA fragments in the environment were identified by functional selection of metagenomic libraries. In a previous study27, we created metagenomic libraries to obtain environmental and clinical resistomes, including (1) river sediment and soil samples from 7 antibiotic-polluted industrial sites in the close vicinity of antibiotic production plants in India73 (anthropogenic soil microbiome), (2) faecal samples from 10 European individuals who had not taken any antibiotics for at least 1 year before sample donation (that is, gut microbiome), and (3) samples from a pool of 68 MDR bacteria isolated in healthcare facilities or obtained from strain collections (Supplementary Table 5). For full details on library construction, see ref. 27.
Briefly, environmental and genomic DNA was isolated using the DNeasy PowerSoil Kit (Qiagen) and the GenElute Bacterial Genomic DNA Kit (Sigma), respectively. Environmental and genomic DNA was enzymatically fragmented, followed by size selection of 1.5–5 kb long fragments. Metagenomic inserts were cloned into a medium-copy-number plasmid and flanked by two 10 nt-long barcodes (referred to as uptag and downtag). Library sizes ranged from 2 to 6 million clones with an average insert size of 2 kb.
Libraries were introduced into K. pneumoniae ATCC 10031 and E. coli ATCC 25922 by bacteriophage transduction (DEEPMINE)27 and electroporation, respectively. DEEPMINE uses hybrid T7 bacteriophage-transducing particles to alter phage host specificity and efficiency for functional metagenomics in target clinical bacterial strains.
In this study, we followed previously described protocols with two minor modifications. First, transducing hybrid phages were generated with a T7 phage lacking the gp11, gp12 and gp17 genes, constructed as previously described74. Second, we used a new phage tail donor plasmid for complementing the deleted phage tail genes. This plasmid was cloned using the ΦSG-JL2 phage tail coding genes, the packaging signal region of T7 phage and the pK19 plasmid backbone based on previous work75.
Functional selections for antibiotic resistance were performed on MHB agar plates containing a concentration gradient of the antimicrobial compounds76,77. Cells containing the metagenomic libraries were plated in a cell number covering at least 10× the size of the corresponding metagenomic library. Plates were incubated at 37 °C for 24 h. For each functional selection, a control plate was prepared with the same number of cells containing the metagenomic plasmid without a cloned DNA fragment in its multicloning site. These control plates showed the inhibitory zone of the antimicrobial compound. To isolate the resistant clones from the libraries, sporadic colonies were identified above the inhibitory zone based on the control plate by visual inspection. Colonies were then collected for plasmid isolation (Thermo Scientific GeneJET Plasmid Miniprep Kit). Metagenomic inserts in the resistant hits were sequenced using two complementary sequencing methods. First, random 10 nt barcodes flanking the metagenomic inserts (pZET_bc_F_SrfI_v2, pZET_bc_R; Supplementary Table 7) on the resistant plasmids from each selection experiment were PCR amplified. For this, we used primers that contain 2× 8-nt-long barcodes specific for each selection experiment (with codes starting with Uptag-UF, Uptag-UR, pZET_Down_F and pZET_Down_R; Supplementary Table 7). Amplicons were pooled, size-selected on agarose gel and sequenced by Illumina. Second, metagenomic inserts and their flanking 10 nt uptag and downtag barcodes were sequenced by Nanopore.
Annotation of ARGsConsensus insert sequences from Nanopore sequencing were matched with the respective selection experiment using the data from Illumina sequencing. First, sequencing reads from Illumina sequencing were demultiplexed using the 2× 8-nt-long barcodes specific to the selection experiment, and then the demultiplexed reads were matched with the consensus insert sequences using the random 10-nt-long barcodes specific to the metagenomic inserts. To reduce redundancy and spurious matches, the list of metagenomic contigs were filtered (1) to unique barcodes, keeping barcodes with the highest Nanopore read count; and (2) to contigs that were supported by at least eight Nanopore reads and five Illumina reads. Prediction of ARGs within these contigs was based on ORF prediction using Prodigal v.2.6.3 (ref. 70), followed by searching the annotated ORFs within the CARD and ResFinder databases78,79. Searches were performed using BLASTX from NCBI BLAST v.2.12.0 (ref. 80) with a 10−5e-value threshold and otherwise default settings. ORFs were clustered at 95% identity and coverage using CD-HIT v.4.8.1 (ref. 81) and only 1 representative ORF was kept for each cluster. The inserts were classified based on whether or not any ARGs were found in them, and whether or not at least one of these ARGs was associated with the antibiotic being tested in that particular selection experiment. Close orthologues of the host-specific proteins were excluded from further analyses by performing a BLASTP search of each ORF on host proteomes (https://www.uniprot.org/proteomes/UP000001734, https://www.uniprot.org/proteomes/UP000029103, downloaded on 24 November 2022) and removing each ORF with higher than 80% sequence similarity. The potential origin of the inserts was assessed by searching the Nanopore contigs within the NCBI Prokaryotic RefSeq Genomes database82 using BLASTN from NCBI BLAST v.2.12.0 with default settings and resolving taxids to hierarchical classifications using R83 and the taxizedb package84,85,86.
Catalogue of mobile ARGsA mobile gene catalogue (that is, a database of recently transferred DNA sequences between bacterial species87) was created previously27. Briefly, 1,377 genomes of diverse human-related bacterial species from the Integrated Microbial Genomes and Microbiomes database87 and 1,417 genomes of Gram-negative ESKAPE pathogens from the NCBI RefSeq database were downloaded. Using NCBI BLASTN 2.10.1+ (ref. 80), we searched the nucleotide sequences shared between genomes belonging to different species. The parameters for filtering the NCBI BLASTN 2.10.1+ BLAST results were as follows: minimum percentage of identity, 99%; minimum alignment length, 500; and maximum alignment length, 20,000. Then, to generate the mobile gene catalogue, we compared them with the merged CARD 3.1.0 (ref. 78) and ResFinder (d48a0fe) databases79 using DIAMOND v.2.0.4.142 (ref. 71). Natural plasmid sequences were identified by downloading 27,939 complete plasmid sequences from the PLSDB database (v.2020-11-19)88. Then the representative sequences of the isolated 114 ARG clusters were searched using BLASTN both in the mobile gene catalogue and in natural plasmid sequences with an identity and coverage threshold of 90%. ARGs were considered mobile if they were present in the mobile gene catalogue and/or in natural plasmid sequences.
Detecting ARGs present in human-associated microbiome and human pathogensTo identify close homologues of the ARGs discovered in our functional metagenomic screens, we used GMGCv1 (ref. 33). This extensive, non-redundant database comprises over 2.3 billion unigenes, derived from more than 13,000 metagenomes across 14 major habitats, and includes detailed phylogenetic origin information. We applied a BLASTN89 search to compare the nucleotide sequences of the ORFs from our screens with all unigenes in the GMGCv1, using a stringent identity and coverage threshold of 90%. ARGs were considered to be associated with the human body if they showed sequence homology to unigenes present in at least five samples in at least one of the following environments: human gut, oral cavity, skin, nose, blood plasma or vagina. To further investigate the association of the detected ARGs with human pathogens, we analysed (1) their presence in the clinical metagenomic library, and (2) their phylogenetic relationships to pathogens, specifically focusing on ESKAPE pathogens and those listed in the WHO priority lists (A. baumannii, P. aeruginosa, Enterobacteriaceae, Enterococcus faecium, Staphylococcus aureus, Helicobacter pylori, Campylobacter, Salmonella, Neisseria gonorrhoeae, Streptococcus pneumoniae, Haemophilus influenzae and Shigella) by leveraging species information metadata from the GMGCv1 database for each BLASTN hit.
Detecting ARGs across E. coli phylogroups, host species types and geographic regionsHost type, geographic location and phylogroup were determined for a dataset of 16,272 E. coli genomes in previous work90. The initial complete dataset of 26,881 E. coli genomes was retrieved from the NCBI RefSeq database (in February 2022) and filtered for genomes with complete metadata. Clermont phylogrouping91 was performed in silico using the EzClermont command-line tool92, whereas host and location metadata were retrieved and categorized using the Bio.Entrez utilities from Biopython v.1.77. All genomes were sorted into the following host species categories: human, agricultural/domestic animals and wild animals. This was achieved using regular expressions constructed by manually reviewing text in the ‘host’ field of the biosample data for each accession number. Geographic locations were split into 20 subregions according to Natural Earth data93. A local BLASTP search was performed for this collection of E. coli genomes against a database of the predicted ARG ORFs identified in functional metagenomic screens, using default parameters. ARGs with both 90% amino acid identity and 90% query coverage per subject, and present in no more than 10% of the examined E. coli genomes, were analysed further.
DIvERGE mutagenesisWe performed deep-scanning mutagenesis in the target genes of moxifloxacin, an established topoisomerase inhibitor. The quinolone resistance-determining regions (QRDR)94 of the gyrA and parC genes were subjected to a single round of mutagenesis using DIvERGE in E. coli K12 MG1655 and K. pneumoniae ATCC 10031. A previously described workflow20 was used with minor modifications. Briefly, cells carrying the pORTMAGE311B plasmid (Addgene number 120418) were inoculated into 2 ml LB medium plus 50 μg ml−1 kanamycin and were grown at 37 °C with continuous shaking (250 rpm) for 12 h. From this starter culture, 500 μl stationary-phase culture was propagated in 50 ml of the same fresh medium under identical conditions. Induction was initiated at a fixed population density (OD600 = 0.4) by adding 50 μl of 1 M m-toluic acid (dissolved in 96% ethyl alcohol; Sigma-Aldrich) for 30–45 min at 37 °C. After induction, cells were cooled on ice for 15 min. Next, cells were washed three times with sterile ice-cold ultrapure distilled water. Finally, the cell pellet was resuspended in 800 μl sterile ultrapure distilled water and kept on ice until electroporation.
To perform DIvERGE mutagenesis, the corresponding gyrA and parC QRDR-targeting oligonucleotides were mixed in equimolar amounts. Of the 500 μl oligonucleotide mixture, 2 μl was added to 40 μl electrocompetent cells in 5 parallel samples. The oligonucleotides we used are listed in Supplementary Table 7. After electroporation, the parallel samples were pooled and suspended in 25 ml fresh LB medium to allow for cell recovery (37 °C and 250 rpm). After a 60 min recovery period, an additional 25 ml LB medium was added and cells were grown for an additional 24 h.
To select clones with reduced susceptibility to moxifloxacin, 500 μl of each mutant cell library was spread onto moxifloxacin-containing MHB agar plates. The plates were incubated at 37 °C for 48 h. Finally, 20-20 antibiotic-resistant clones were selected randomly and analysed further by capillary sequencing using the PCR primers listed in Supplementary Table 7.
Efflux activity measurementsMeasuring the accumulation of the fluorescent Hoechst dye is known as a robust and rapid method for monitoring efflux activity/membrane permeability in bacteria95. This method is based on the intracellular accumulation of the fluorescent probe Hoechst 33342 (Bisbenzimide H 33342; Sigma-Aldrich). Cells were grown overnight in MHB, then 20 µl of the overnight culture was used to inoculate 2 ml of MHB liquid medium and then the cells were grown to mid-exponential phase (OD600 = 0.4–0.6). Bacterial cultures were harvested by centrifugation at 4,500g for 30 min. Next, cells were washed and resuspended in the buffer containing 5 mM HEPES (pH 7.0) and 5 mM glucose. The OD600 of the cell suspensions was adjusted to 0.1, and 0.18 ml of each suspension was transferred to 96 well plates (CellCarrier-96 Black Optically Clear Bottom; Sigma-Aldrich). Plates were incubated in a Synergy H1 microplate reader at 37 °C and 25 μM Hoechst 33342 was added to each well. The ancestor strain was treated with an efflux inhibitor agent (phenylalanine-arginine β-naphthylamide) that served as a positive control. The OD600 and fluorescence curves were recorded for 2 h with 75 s delays between readings and 2.5 min reading intervals. Fluorescence reading was performed from the top of the wells using excitation and emission filters of 355 nm and 460 nm, respectively. To estimate changes in efflux activity, we used a 2 step process: (1) we measured the optical-density-normalized fluorescence signal over a fixed time frame (from 7.5 min to 120 min) to monitor the intracellular accumulation of a fluorescent probe, and (2) we calculated the change in normalized fluorescence signal by dividing the signal at the final time point (120 min) by that at the initial time point (7.5 min). Relative efflux activity of the tested strains was determined by normalizing the reached raw values to those of their respective ancestral strains and taking its inverse.
Reporting summaryFurther information on research design is available in the
留言 (0)