Cell culture, cloning, transfection, and immunofluorescence
Human fibroblasts were grown a described previously [6]. Human embryonic Kidney 293 T (HEK293T) cells were cultured in DMEM with 10% FBS. HEK293T cells were transfected using standard protocols for Lipofectamine 3000 (Thermo Fisher Scientific, Germany). All experiments were performed 48–72 h after transfection. Generation of expression plasmids for the ANO3 variants A599D, S651N, V561L, and S116L was described in our previous report [6]. For patch clamp experiments and intracellular calcium measurements, ANO3-cDNA was subcloned into the bicistronic vector pIRES1-CD8. Immunofluorescence of overexpressed ORAI1 was performed with a mouse monoclonal anti-hORAI1 #168 and a donkey anti-mouse Alexa 488 (Sigma-Aldrich, Taufkirchen, Germany).
Measurement of intracellular Ca2+ concentrations
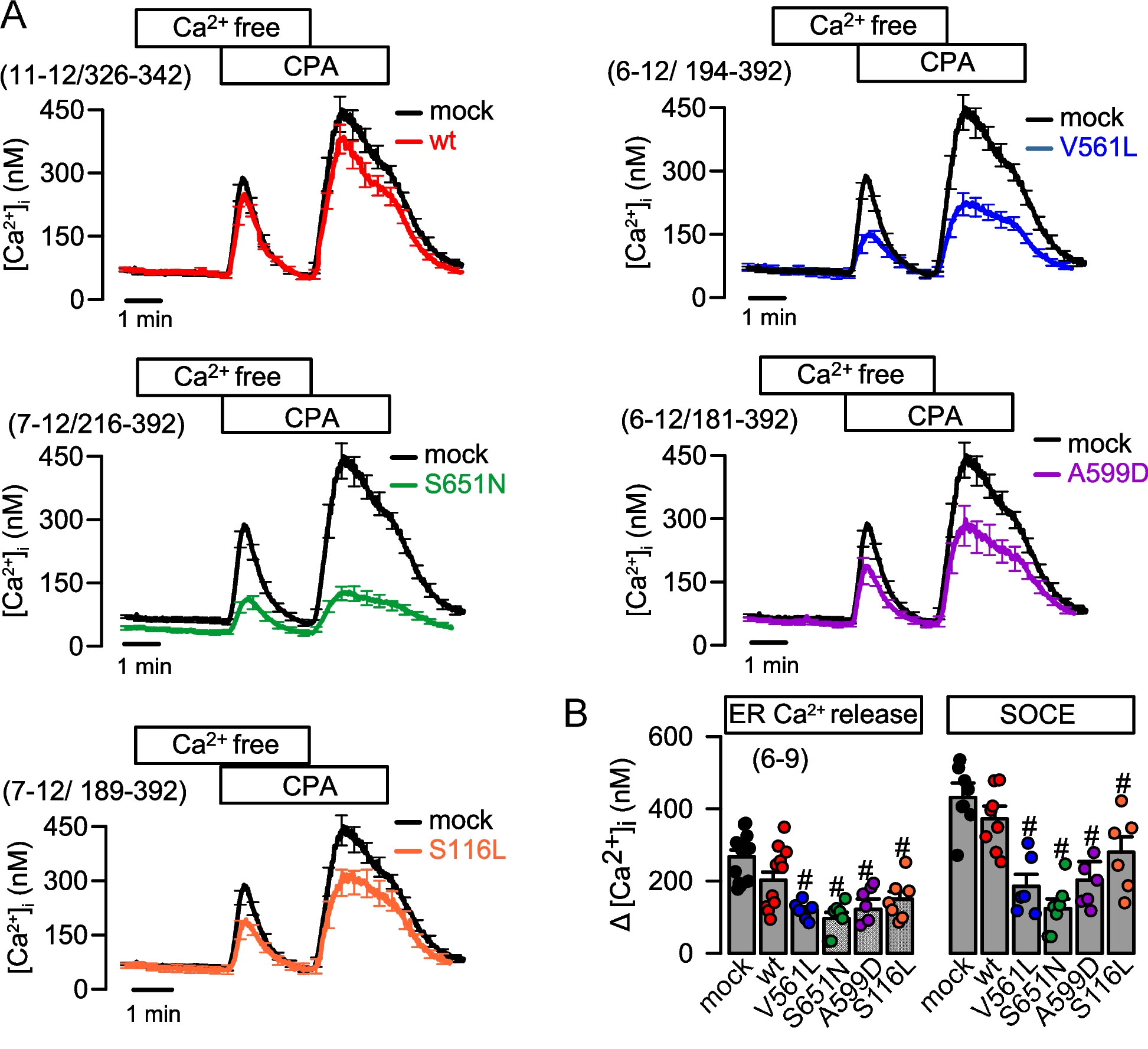
Measurements of cytosolic Ca2+ were performed as described recently [7]. In brief, cells were loaded with 2 µM Fura-2, AM (BIOZOL, Eching, Germany) in OptiMEM (Gibco, Thermo Fisher, Scientific, Waltham, MA 02451, USA) with 0.02% Pluronic F-127 (Invitrogen, Thermo Fisher, Scientific, Waltham, MA 02451, USA) in Ringer solution (mmol/l: NaCl 145; KH2PO4 0,4; K2HPO4 1,6; Glucose 5; MgCl2 1; Ca2+-Gluconate 1,3) for 1 h at room temperature. Fluorescence was detected in cells perfused with Ringer’s solution at 37 °C using an inverted microscope (Axiovert S100, Zeiss, Germany) and a high-speed polychromator system (VisiChrome, Puchheim, Germany). Fura-2 was excited at 340/380 nm, and the emission was recorded between 470 and 550 nm using a CCD-camera (CoolSnap HQ, Visitron Systems, Germany). [Ca2+]i was calculated from the 340/380 nm fluorescence ratio after background subtraction. The formula used to calculate [Ca2+]i was [Ca2+]i = Kd x (R-Rmin)/(Rmax-R) x (Sf2/Sb2), where R is the observed fluorescence ratio. The values Rmax and Rmin (maximum and minimum ratios) and the constant Sf2/Sb2 (fluorescence of free and Ca2+-bound Fura-2 at 380 nm) were calculated using 2 µmol/liter ionomycin (Biomol GmbH, Hamburg, Germany) and 5 mmol/liter EGTA to equilibrate intracellular and extracellular Ca2+ in intact Fura-2-loaded cells. The dissociation constant for the Fura-2•Ca2+ complex was taken as 224 nmol/liter [8]. Control of the experiment, imaging acquisition, and data analysis were done with the software package Meta-Fluor (Universal Imaging, USA).
Patch clamping
Cells were patch clamped when grown on fibronectin-coated glass coverslips at 37 °C. Patch pipettes were filled with a cytosolic-like solution containing (in mM): KCl 30, K-Gluconate 95, NaH2PO4 1.2, Na2HPO4 4.8, EGTA 1, Ca-Gluconate 0.758, MgCl2 1.03, D-Glucose 5, ATP 3; pH 7.2. The intracellular (pipette) Ca2+ activity was 0.1 µM. The bath was perfused continuously with Ringer’s solution (in mM): NaCl 145, KH2PO4 0.4, K2HPO4 1.6, Glucose 5, MgCl2 1, and Ca-Gluconate 1.3) at a rate of 6 mL/min. Patch pipettes had an input resistance of 2–5 MΩ and whole cell currents were corrected for serial resistance. The current voltage (I/V) relationships were determined by pulsing from the holding potential of −100 mV to test potentials between −100 and + 100 mV, increasing in 20 mV increments. Currents were recorded using the EPC-9 computer-controlled amplifier, PULSE software (HEKA), and Chart software (AD Instruments). The current density was calculated by dividing whole cell currents by cell capacitance.
Cell death assays, flow cytometry
Cells were collected using accutase (Capricorn Scientific, Ebsdorfergrund, Germany), washed with cold Dulbecco’s PBS (DPBS), and centrifuged at 500 g and 4 °C for 10 min. Subsequently, cells were resuspended in 100 μL annexin binding buffer containing 5 μL annexin V-FITC and 2.5 μL 7-aminoactinomycin D (7-AAD; BioLegend, Koblenz, Germany). Cells were incubated with 10 μM ionomycin for 20 min. Reactions were stopped by adding 400 μL of DPBS, and cells were analyzed immediately. Fluorescence-activated cell sorting (FACS) analyses were performed in Annexin V standard binding buffer (BioLegend, San Diego, CA, USA) containing 10 mM Hepes, 140 mM NaCl, and 2.5 mM CaCl. For each measurement, at least 10,000 cells were analyzed by flow cytometry at 37 °C (BD AccuriTM C6, St. Ives, UK) 7-AAD, a non-permeant dye, was used to identify cells with plasma membrane leakage. Fibroblasts were labelled by propidium iodide uptake.
Biotinylation
Biotinylation was described in our previous report [9]. After biotinylation for 30 min at 4 °C, cells were collected and lysed in 0.5% NonidetP40 lysis buffer. Proteins were separated by 8.5% SDS-PAGE and transferred to a PVDF membrane. The membrane was blocked with 5%NFM/TBS-T or 5%NFM/PBS-T for 1 h and incubated overnight at 4 °C with mouse anti-ORAI1 (diluted 1:1000 in 1% BSA/TBS-T; Sigma, Taufkirchen, Germany). Immunoreactive signals were detected using the SuperSignal chemiluminescence substrate (Pierce, Waltham, USA).
Statistical analysis
Statistical analysis was performed using Student’s t-test (for paired or unpaired samples as appropriate) or ANOVA. A value of p < 0.05 was accepted as a significant difference. Data are reported as means ± SEM.



留言 (0)