Data collection and preprocessing
Stomach adenocarcinoma (STAD) is the most common pathological type of GC in clinic, accounting for about 95% of GC patients. Therefore, this study focuses on STAD as the research object. The processed original mRNA expression data for The Cancer Genome Atlas Stomach Adenocarcinoma (TCGA-STAD) data, including the normal group (n = 36) and the tumor group (n = 412), were downloaded from the Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/). 161 ARGs used in this analysis were obtained from the GSEA database (https://www.gsea-msigdb.org), which mediating programmed cell death by activation of caspases (Human Gene Set: HALLMARK_APOPTOSIS).
Consistent clustering analysis based on ARGs
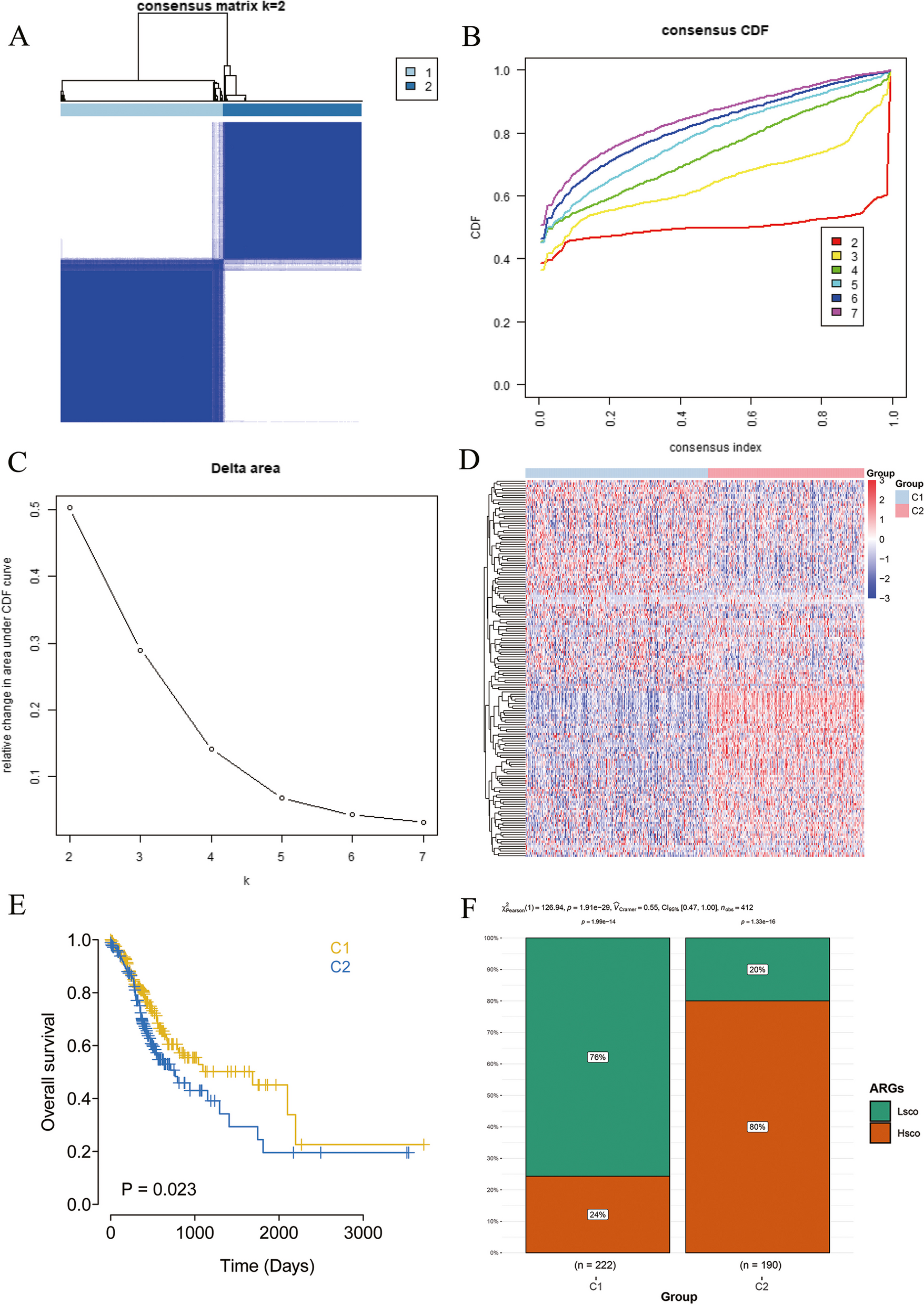
Based on the expression levels of candidate gene sets, GC samples were clustered using the consistent clustering function. The clustering was repeated 50 times, with each iteration including 90% of the samples. The optimal number of clusters was determined based on the cumulative distribution function curve of the consistency score, and the characteristics of the consistency matrix heat map. When the consistency score is the highest and there is a significant difference in OS between subtypes, the clustering number k is the optimal number.
Immune cell infiltration
The Microenvironment Cell Population (MCP) algorithm was used to evaluate eight immune and two non-immune stromal cell populations (immune cell populations: T cells, CD8+ T cells, natural killer cells, cytotoxic lymphocytes, B cell lineage, monocyte lineage cells, myeloid like dendritic cells and neutrophils; stromal cell populations: endothelial cells and fibroblasts). In addition, another method used in this study to estimate immune infiltration was single sample gene set enrichment analysis (ssGSEA), which calculates an enrichment score that indicates the degree of coordinated upregulation or downregulation of genes in a specific gene set in a single sample. The Gene Set Variation Analysis (GSVA) R package was used to estimate six additional immune cell populations, including regulatory T cells (Treg), helper T cells 1 (Th1), helper T cells 2 (Th2), helper T cells 17 (Th17), central memory T cells, and effector memory T cells (Tem). Additionally, the ESTIMATE algorithm was used to calculate immune and stromal scores, which can reflect the abundance of gene features related to stromal and immune cells.
GSEA analysis of GC subtypes based on ARGs
The log2(FoldChange) value of each gene between subtypes was obtained using the limma package. GSEA analysis of Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways was then performed using the R package clusterProfiler. Finally, the top 10 upregulated pathways with the highest normalized enrichment score (NES) values were selected for each subtype. The gene set was obtained from the Molecular Signatures Database (MSigDB) (http://www.gsea-msigdb.org/gsea/downloads.jsp).
Performance validation of GC subtypes based on ARGs
The LIMMA package was used to analyze the differentially expressed genes (DEGs) between GC subtypes, with a corrected P value of less than 0.05 as the screening criteria. Only genes with significant differential expression in all possible comparisons were considered as subtype-specific genes. Further, the top 30 genes with the largest log2FC values in each subtype were selected to build a prediction model and generate 60 gene classifiers. The nearest template prediction (NTP) algorithm was then used to predict subtypes using the 60 genes on the dataset and compared with the classification results based on the NTP algorithm.
Immune therapy and drug sensitivity analysis
The similarity between the gene expression profiles of our subtypes and those of previously published samples treated with immunotherapy was measured using the SubMap analysis, indirectly predicting the effectiveness of immunotherapy for our subtypes. In addition, the Genomics of Drug Sensitivity in Cancer (GDSC) (https://www.cancerrxgene.org/) was used to predict the chemotherapy sensitivity of each tumor sample by employing the R package “pRRophetic”. IC50 estimates for each specific chemotherapy drug were obtained using regression and validated using 10-fold cross-validation on the GDSC training set. All parameters were selected with default values, including “combat” for batch effect removal and averaging replicate gene expression values.
Analysis of differences in tumor microbiome among GC subtypes based on ARGs
The data was collected from a study that used the Kraken analysis to investigate the tumor microbiome [15]. Further prediction and validation of subtypes were performed by analyzing the interactions between immune infiltration-microbiome, gene expression-microbiome and immune therapy-microbiome, with genes including immune genes, cytotoxic T lymphocytes (CTL) genes and key genes [16].
Gene function enrichment analysis
Functional annotation of important gene sets was performed using the Metascape database (www.metascape.org) to comprehensively explore the functional relevance of the gene set. GO analysis and KEGG pathway analysis were conducted for specific genes. Min overlap ≥ 3 and P ≤ 0.01 were considered statistically significant.
Gene Set Variation Analysis (GSVA) for key genes
In this study, gene sets were downloaded from the Molecular Signatures Database (version 7.0), and the GSVA algorithm was used to comprehensively score each gene set and evaluate potential biological functional changes in different samples.
Gene Set Enrichment Analysis (GSEA) for key genes
GSEA (http://www.broadinstitute.org/gsea) was performed on the expression profiles of GC patients to identify differentially expressed genes between the high expression group and the low expression group. Gene sets were filtered based on a maximum and minimum size of 500 and 15 genes, respectively. After 100 permutations, significantly enriched gene sets were obtained based on P-value < 0.05 and False Discovery Rate (FDR) value of 0.25.
Cell culture and cell transfection
Human GC cell lines (AGS, HGC-27, and NUGC-3) along with the normal gastric mucosal epithelial cell line GES-1 were obtained from the Cell Bank of the Chinese Academy of Sciences or Shanghai Institute of Biochemistry and Cell Biology. These cells were maintained in RPMI-1640 medium (Invitrogen, Grand Island, USA) supplemented with 10% fetal bovine serum (FBS) and incubated at 37°C with 5% CO2. Small interfering RNAs (siRNAs) targeting GPX3 (si-GPX3), CAV1 (si-CAV1), PLAT (si-PLAT) and their corresponding negative control (si-NC) were designed and synthesized by Shanghai GenePharma Co., Ltd. The sequences of the siRNAs used were as follows: 5ʹ-UAGAUGGAAUAGACACGGCTT-3ʹ (si-CAV1), 5ʹ-AUGUCCAUCUUGACGUUGCTT-3ʹ (si-GPX3), 5ʹ-UUAAAGACGUAGCACCAGGTT-3ʹ (si-PLAT), and 5ʹ-ACGUGACACGUUCGGAGAATT-3ʹ (si-NC).
Cells were seeded in 6-well plates. After cells reached approximately 40–60% confluence, cells were transfected with siRNA (100nM) or si-NC (100nM) using Lipofectamine 2000 reagent (Invitrogen, CA, USA) and Opti-MEM I Reduced Serum Medium (Gibco, CA, USA) in accordance with the manufacturer’s instructions, respectively. After 5 h of cell transfection, culture medium was replaced by RPMI-1640 medium supplemented with 10% FBS.
Clinical specimen collection
STATBOX (https://www.trialstats.com/statbox/), online sample size calculation software, was used in sample size calculation for clinical validation. A total of 37 paired gastric cancer tissues and adjacent normal tissues were collected from surgical patients at the First Affiliated Hospital of Ningbo University from March 2022 to September 2023. None of the patients had undergone radiotherapy or chemotherapy prior to surgery, and all cancer tissues were confirmed by pathological examination. This study received approval from the ethics committee of the First Affiliated Hospital of Ningbo University (IRB No. KY20220101). Written informed consent was obtained from all patients.
RNA extraction and PCR detection
RNA was extracted using TRIzol (Ambion, Carlsbad, USA) and reverse transcribed to cDNA with a GoScript Reverse Transcription (RT) System (Promega, Madison, USA). Subsequently, PCR was carried out using GoTaq qPCR master mix (Promega). The cycling parameters included predenaturation at 95°C for 5 min, denaturation at 94°C for 15 s, annealing at 53°C for 30 s, extension at 72°C for 30 s, repeated for 40 cycles. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as the control. Primer sequences were designed for GPX3, CAV1, PLAT, and GAPDH in Supplementary Table 1. ΔCt method was employed for quantification, where a higher ΔCt value indicates a lower expression level.
Cell proliferation assay
After cell transfection for 24 h, cells were seeded in 96-well plates at a density of 2 × 103 cells/well. After incubation of 0 h, 24 h, 48 h, 72 h, 96 h, 10 µl CCK-8 reagent (Beyotime, Shanghai, China) was added to each well and incubated at 37°C for 2 h. The optional density (OD) of each well at 450 nm was detected by the multifunctional microplate reader (Thermo Fisher Scientific, MA, USA). Each sample had six replicates.
Plate colony formation assay
After cell transfection for 24 h, cells were trypsinized to single cell suspensions and then seeded into 6-well plates at a density of 600 cells/well. After 10 days, each well was washed with PBS for two times, fixed with 4% paraformaldehyde (Solarbio, Beijing, China) for 30 min and then stained with 0.1% crystal violet staining solution (Solarbio) for 30 min. Finally, each well was photographed and the number of stained colonies was counted. Each assay was repeated three times.
Transwell invasion assays
Transwell invasion assays were performed using transwell chambers (Corning, NY, USA) with 8.0 μm-pore polycarbonate membranes paved with matrigel mix (100 µg/ml) (BD, NJ, USA). After cell transfection for 24 h, cells were diluted to 1 ~ 1.5 × 105/mL with serum-free RPMI 1640 medium. The bottom chamber was filled with 500 µL of RPMI 1640 medium supplemented with 20% FBS, whereas the upper chamber was added to 200 µL of cell suspension. After incubation for 36 h at 37°C, the cells on the membrane of the lower surface were washed with PBS for three times, fixed with paraformaldehyde for 30 min, and stained with 0.1% crystal violet solution for 30 min. Each assay was repeated three times.
Statistical analysis
Statistical analyses in this study were conducted using R software (version 4.4.0) (https://www.r-project.org/) and Statistical Product and Service Solutions (SPSS) 19.0 software. Graphs were created using R software and GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA). All statistical tests with statistical significance defined as P < 0.05.


留言 (0)