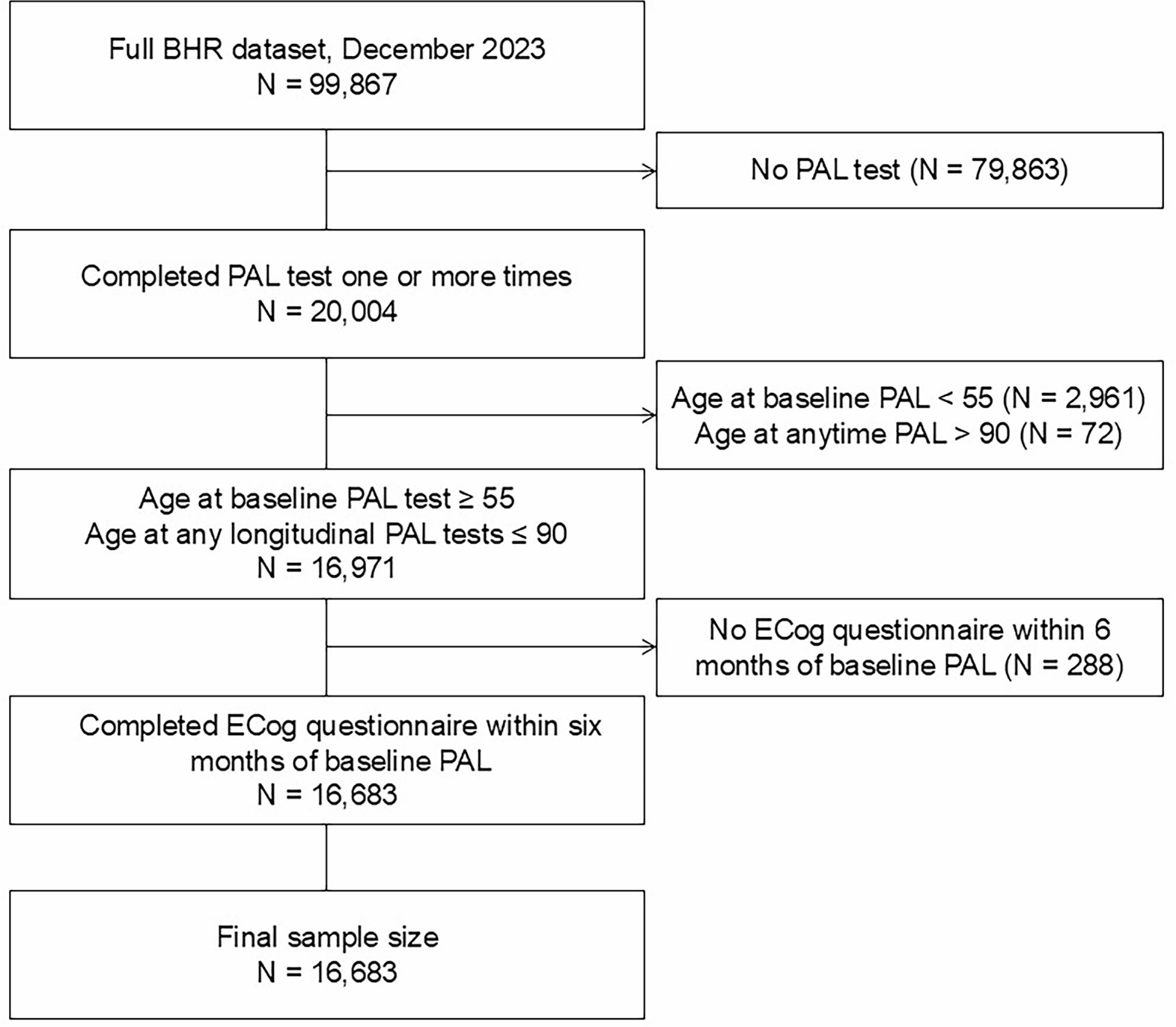
Individuals included in the study
The presence of IRIS-1 and 2 was analyzed in Cornu Ammonis 1 (CA1) of postmortem collected hippocampal samples from cases (Netherlands Brain Bank; NBB) separated into three groups. Group 1 A (n = 6) included (n = 3) non-demented controls (NC) and (n = 3) clinically diagnosed and neuropathologically verified AD cases, Group 1B included (n = 7) clinically diagnosed and neuropathologically verified AD cases, and Group 2 (n = 6) consisted of (n = 3) non‐demented controls without T2D (NC) and (n = 3) clinically diagnosed non-demented T2D cases (NC-T2D). The demographics of the individuals are shown in Table 1. The presence of NFT and LB was scored according to Braak stages I-VI [11] and the Aβ plaques were scored into O, A, B, C, where O = zero, A = some, B = moderate and C = many [11]. Cases without cognitive impairments and NFT scores below III and Aβ below A were considered as NC.
Ethical approval and patient consents
Informed consent for the use of brain tissue, plasma, and clinical data for research purposes were obtained from all subjects or their legal representatives in accordance with the International Declaration of Helsinki [15]. The tissue collection protocols were approved by the medical ethics committee of VU medical Centre in Amsterdam, the Netherlands and the Swedish Ethical Review Authority approved the study (Dnr 2016/155, 2017/717).
Brain sample preparation
Directly after autopsy, brain samples containing hippocampus and entorhinal cortex in Group 1 A and B were immersion-fixed in paraformaldehyde (PFA) (4%) for 14–20 h. Brain samples in Group 2 were freshly frozen without cryoprotectant directly after autopsy and remained frozen until immersion-fixed in PFA (4%) for 4 h. After fixation, the samples from all groups were incubated in phosphate-buffered saline (PBS) with 30% sucrose. They were then sectioned in 40 μm free-floating sections and stored in cryoprotectants at − 20 °C until they were used for immunostaining.
Brain samples immunostaining
For analysis of cellular localization of IRIS-1 and 2 sections from NC and AD (Group 1 A) were stained with antibodies against IRIS-1 and 2 (in-house generated rabbit antibodies against the unique C-terminal peptides existing in the novel isoforms but not in canonical CD59 [10]), together with antibodies directed against Iba-1 (microglia marker), GFAP (astrocyte marker) and NeuN (neuronal marker). The sections were incubated for 1 h with blocking solution containing 5% goat serum (Jackson Immunoresearch) and 0.25% Triton X-100 in PBS, and then incubated with rabbit-anti-IRIS-1 or rabbit-anti-IRIS-2 (Capra, #613.610 and #612.609 respectively) together with either chicken-anti-GFAP (1:500, Merck) or Mouse anti-NeuN (1:200, Proteintech) in blocking solution overnight (ON) at 4 °C. The next day the sections were washed and incubated with secondary antibodies Alexa 488 goat-anti-mouse (#A11029, Thermo Fisher Scientific) and dylight 549 goat-anti-rabbit (#DI-1549, Vector Laboratories) in blocking solution for 2 h at room temperature (RT). The Iba-1 co-staining with IRIS-1 and 2 was performed sequentially, starting with the staining against IRIS-1 or 2 according to the above instructions. The day after the sections were incubated with rabbit anti-Iba-1 (1:500, Wako) ON at 4 °C and with Alexa 488 goat-anti-rabbit (#A11029, Thermo Fisher Scientific) for 2 h the following day. To analyze the relationship between p-tau load and IRIS-1 and 2, sections (Group 1B) were stained against AT8 (phosphorylation of tau at sites Ser202, Thr205) and IRIS-1 or 2. The same protocol as above was used with mouse-anti-AT8 (1:500, Invitrogen) and the IRIS-1 and 2 antibodies as the primary antibody. Sections stained according to all the above immunofluorescence protocols, but with PBS instead of the primary antibodies were used as negative control. All stained sections were incubated in Sudan Black (1% in 70% ethanol) (Sigma-Aldrich) for 5 min before they were rinsed and mounted with a Vectashield Set mounting medium containing DAPI (Vector Laboratories).
For analysis of IRIS-1 and IRIS-2 expression in hippocampus and entorhinal cortex and quantification of the mean signal intensity of IRIS-1 and IRIS-2 immunoreactivity, sections from NC and AD (Group 1 A), NC, and T2D-NC (Group 2) were immunohistochemically stained with antibodies against IRIS-1 and 2. The sections were incubated in quenching solution (3% H2O2, 10% methanol) for 30 min, followed by Impress reagent kit blocking solution (Vector Laboratories #MP-7402) for 1 h at room temperature and then incubated with rabbit-anti-IRIS-1 or rabbit-anti-IRIS-2 in blocking solution ON at 4 °C. The following day the sections were incubated in Ig Impress reagent kit secondary anti-rabbit antibody (Vector Laboratories #MP-7401) at RT for 2 h, and then developed for 2 min in 0.25 mg/ mL diaminobenzidine and 0.012% H2O2. The sections were mounted with DPX (Sigma Aldrich, #06522).
Analysis of IRIS-1 and 2 expression in NC, AD and T2D brains
To analyze the mean signal intensity of IRIS-1 and 2 immunoreactivity in Group 1 A, 1B and Group 2 at least 6 images were taken per case with 10–20 neurons per image. Fiji program was used for the quantifications, where a manual threshold was adjusted for each case, and region of interest (ROI) (drawn around single neurons) was selected. Around 10–12 neurons per photo were quantified where the mean value for the mean signal intensity was taken, and compared between three controls and three AD, or non-demented T2D cases. All images and quantifications were captured/ performed blindly.
Analysis of p-tau and IRIS-1 and 2 expression (confocal microscopy)
Sections stained against AT8, IRIS-1 and IRIS-2 were analyzed using confocal microscopy. Z-stack images were taken and the maximum intensity projection was used to compile the stacks into single images using the Zen software. The images were exported as TIFF and the channels for IRIS-1/ 2 and p-tau were then quantified individually for all CA1 neurons with the Fiji software using a ROI (neurons). The ratio of IRIS-1/ 2 to p-tau was taken and categories were made according to the ratio from low, intermediate to high where < 2 was low, 2–4 intermediate and > 4 high for IRIS-1 with 0.5 lower threshold for all categories for IRIS-2. A minimum of 10 neurons per ratio category were quantified per case (n = 7). Due to having clustered data where neurons between cases cannot be considered independent, a linear mixed model was used to avoid pseudoreplication. To analyze the data statistically, R 4.3.3. was used with the Ime4 package. The following model specifications were used: Imer (IRIS-1/ 2 ∼ ptau + (1| case), REML = FALSE, control = lmerControl (‘bobyqa’), data = MixedModel). A likelihood ratio test (LRT) was used to see if the full model was significantly better than the null model (Alfa = 0.05), and the 95% confidence interval was extracted to get a sense of the variation. The model assumes that the residuals are normally distributed, which was assessed by plotting them against a normal distribution curve. The LRT assumes a Chi-square distribution, and that the restricted (residual) maximum likelihood (REML) is set to false. We assumed fixed slopes and random intercepts for the linear mixed model since that is the standard, and we had no theoretical reason to believe otherwise. The control used was BOBYQA, which performs a derivative-free bound-constrained optimization using an iteratively constructed quadratic approximation for the objective function, which is used to optimize the models performance.
Localization of IRIS-1 and 2 in astrocytes, microglia and neurons
Localisation of IRIS-1 and IRIS-2 in microglia (Iba-1 positive cells)) and astrocytes (GFAP-positive cells) and neurons (NeuN) were analyzed by capturing Z-stack images, which were compiled into a single image using the maximum intensity projection in Zen software, and thereafter adjusted for clarity. For analysis of IRIS-1 and 2 in astrocytes and microglia at least 10 images were taken from three cases in Group 1 A (in total n = 30), while the presence of IRIS-1 and 2 in neurons were analyzed in one NC case.
Table 1 Clinical diagnosis, sex, age, neuropathological assessment, cause of death and postmortem delay of cases included in the studySH-SY5Y cell line
SH-SY5Y cells is a neuroblastoma cell line derived from a 4-year-old female patient with metastatic bone tumor [16]. Cells were regularly tested for Mycoplasma (Eurofins Genomic, detection based on Sanger sequencing), and were cultured in a humidified chamber with 5% CO2 at 37 °C. SH-SY5Y cells can be differentiated into mature neuron-like phenotype characterized by presence of neuronal markers using BDNF and retinoic acid. In brief, SH-SY5Y cells (Sigma-Aldrich, St. Louis, MO, USA) were cultured in Eagle’s minimum essential medium (EMEM, Sigma, #M2279), and Ham’s F-12 nutrient mix (Sigma, #N4888), supplemented with nonessential amino acids (Sigma, #M7145), 15% fetal bovine serum (ATCC), 500U L-Glutamine (Cytiva), penicillin (100 U/ ml) and streptomycin (100 µg/ ml). Cell culture plates were coated with 10 g/ ml of poly-D-lysine, 1 h prior to cells seeding.
SH-SY5Y cells differentiation into mature neurons
Culture medium was replaced 1–2 days after seeding with phenol free Dulbecco’s Modified Eagle Medium (DMEM), supplemented with 5% fetal bovine serum (ATCC) and 10 µM all-trans-retinoic acid (ATRA). Cells were grown in an ATRA-containing medium for 3 days and the medium was refreshed every day. The medium was replaced with a second differentiation medium, containing phenol-free neurobasal medium (containing GlutaMAX, 200 mM), 1x N2-supplement and 50 ng/ml brain-derived neurotrophic factor (BDNF). Cells were grown in BDNF-containing neurobasal medium for a minimum of 4 days, and the medium was refreshed daily. Differentiation was monitored microscopically via morphological assessment of neurite outgrowth, as well by real-time PCR using the neural markers.
RT-PCR
RNA was isolated from differentiated and non-differentiated SH-SY5Y cells using RNA purification kit (Qiagen, #74136). cDNA was synthesized using oligo-dT primers and Superscript IV (Invitrogen) according to standard protocol, and amplicon amplified using DreamTaq Green PCR Master mix (Thermo Scientific) using annealing temperature 62 °C for 30 s for CD59, IRIS-1 and IRIS-2, and 54 °C for 30 s for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). For all the amplicons extension time of 30 s was used, with final extension of 15 min. Samples were separated on 1.2% agarose gel and visualized using SybrSafe reagent. GAPDH was used as a control. For detection of total human CD59 transcript primers 1 and 2 were used. To detect human IRIS-1, primers 1 and 3 were used, and to detect human IRIS-2, primers 4 and 2 were used.
Primer 1: Forward; ATC ACA ATG GGA ATC CAA GGA GGG.
Primer 2: Reverse; CTC TCC TGG TGT TGA CTT AGG G.
Primer 3: IRIS-1 Reverse; CTC AGG AGA GAG AGG CCG AC.
Primer 4: IRIS-2 Forward; AGT TGG GAT ATC ACT ATG TTG CCC.
Semi-quantitative RT-PCR
The cDNA was amplified with TaKaRa Ex Premier™ DNA polymerase 2x PCR Master mix (TaKaRa) with the same programs for IRIS-2/CD59 and GAPDH but with an extension temperature of 68 °C, a denaturation temperature of 98 °C for 10 s, and a final extension of 5 min. For IRIS-1, the annealing temperature was 57 °C for 10 s. Samples were separated on 1.1% agarose gels and visualized using SybrSafe reagent.
Western blots
Cells were lysed in RIPA lysis buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.5, 1% NP-40, and 0.5% deoxycholate) with addition of protease and phosphatase inhibitors (Thermo Scientific). Proteins were resolved on 15% SDS-PAGE gel (home-made), or 4–20% Precast Protein gels (Bio-Rad) under reducing conditions unless specified otherwise, transferred onto PVDF membrane using Trans-Blot Turbo system (Bio-Rad) and blocked with Quench buffer (3% fish gelatin in Immunowash: 50 mM Tris-HCl, 150 mM NaCl, 0.1% Tween 20, pH 8.0), 5% BSA in immunowash (Sigma-Aldritch #126579), or EveryBlot (Bio-Rad). The membranes were probed with suitable antibodies followed by the addition of appropriate HRP-conjugated secondary antibody (Dako) specifically stated in each subsection, and development with ECL reagent (Santa Cruz Biotechnology).The membranes were imaged using a CCD camera (Bio-Rad) and analyzed in Image Lab.
Effect of glucolipotoxicity on IRIS-1/2 expression in SH-SY5Y cells
Cells were seeded in 12-well plates (2 × 105 cells/ well) or 24-well plates (1.5 × 105 cells/ well) for IRIS-1 and IRIS-2 analysis, respectively. The cells were then treated for 48 h with Opti-MEM medium supplemented with various combinations of 25 mM glucose, 200 µM palmitic acid, 50 or 100 ng/ ml TNF-α ImmunoTools), and 100 ng/ ml IFN-γ (ImmunoTools). Medium was replaced after 24 h. The control wells were untreated, containing Opti-MEM medium only. For the Western blot, the antibodies used for IRIS-1 was Capra, custom made #614.610 or #613.610, 1:1000 in Quench, with the goat-anti-rabbit Abcam/Dako 1:5000. For IRIS-2 the Capra, custom made #612.609 antibody was used, 1:1000 in Quench with Dako goat-anti-rabbit 1:5000. For the loading control anti-β-tubulin, 1:25000 in Quench (Abcam #ab6046) was used, with the Dako goat-anti-rabbit 1:25000.
qRT-PCR
RNA was extracted using RNeasy plus Mini kit (Qiagen) and cDNA was synthesized using oligo-dT primers and Superscript IV (Invitrogen). Quantitative PCR was performed with specific TaqMan probes (Applied Biosystem) and Viia7 Real-Time PCR system (Thermo Fisher). Expression levels of CD59, CDK5, microtubule-associated protein tau (MAPT), GSK3β, and ADAM17 (Thermo Scientific) were calculated after normalization with the mean of the housekeeping gene GAPDH. 2-ΔΔCt method was used to determine the gene expression differences among the groups. For the standard PCR, the gels were imaged directly after electrophoresis using a CCD camera (Bio-Rad). The band intensities were then quantified from the ratio of IRIS-1/2 and CD59 to GAPDH in Image Lab (Bio-Rad).
Noradrenaline secretion ELISA
SH-SY5Y cells were seeded on a 24-well plate. Knockdown of CD59, IRIS-1 and IRIS-2 was performed by incubation with siRNA targeting CD59, 300 nM (Ambion #4392422) and 1.5 µl lipofectamine RNAiMax (Invitrogen) for 72 h. Non-targeting negative control siRNA (Ambion, #4390843) was used as a negative control. After that time the cells were washed 1x with PBS and incubated for 30 min. at 37 °C with HEPES-buffered saline (HBS) (0,44 mM KH2PO4, 1,2 mM MgCl2, 2 mM CaCl2, 4,2 mM NaHCO3, 20 mM HEPES, 137 mM NaCl, 5 mM KCl, 5 mM glucose, 0.2 mM pargyline, 0.2 mM ascorbic acid, pH 7,4). Noradrenaline secretion was measured by incubating the cells for 30 min with HBS containing 5 mmol/L KCl (basal release), or with HBS containing 100 mmol/L KCl (evoked release). The supernatants were collected. Cells were washed 3x with ice-cold PBS, and lysed with a RIPA buffer containing protease and phosphatase inhibitors. The total protein content was measured using BCA kit (Thermo Scientific). Further, the noradrenaline level in the supernatant was determined and the cell extracts were used for total protein quantitation. The noradrenaline ELISA was performed according to the manufacturer instructions (EKN47439, 96 tests). Additionally, the CD59, IRIS-1 and IRIS-2 knockdown efficiency was verified by Western Blotting. The antibodies used were Capra #614.610 or #613.610 custom made antibody for IRIS-1, 1:1000 in EveryBlot/Quench with secondary Abcam/Dako 1:5000 goat-anti-rabbit. For IRIS-2 the Capra #612.609 custom made antibodies were used 1:1000 in Quench, with Dako goat-anti-rabbit 1:5000. For CD59 the BRICK 229 (IBGRL research #9409P) antibody was used, 1:2000 in 5% milk in immunowash, with Dako goat-anti-mouse 1:5000 as secondary antibody. For the loading control, anti-β-tubulin (Abcam #Ab6046) was used 1:25000/1:10000 in Quench with Dako goat-anti-rabbit 1:25000/1:10000.
Effects of CD59 knockdown on p-tau and Cdk5 expression
Knockdown of CD59 was performed and its efficiency verified in the same way as described in the “Noradrenaline secretion assay” methods section. Western blotting was used to analyse the effects of CD59 knockdown on p-tau and Cdk5 expression. Membranes were probed with the following antibodies: anti-Cdk5 (1:1000, Abcam #2506) in Quench buffer, non-reduced conditions, Dako goat-anti-rabbit 1:2000, anti-Tau (1:1000, Invitrogen #AHB0042) in 5% BSA with Dako goat-anti-mouse 1:2000, AT8 (Ser202/ Thr205, 1:1000, Invitrogen #MN1020) in 5% BSA, with Dako goat-anti-mouse 1:2000, and anti-GAPDH (Abcam #Ab8245) 1:10000 in Quench buffer with Dako goat-anti-mouse 1:10000.
Subcellular fractionation
SH-SY5Y cells were seeded on and harvested from a petri dish by trypsinization (5 × 106 cells). Cells were then fractionated using MEM-PER Plus Kit (Thermo Scientific, #89842) according to the manufacturer’s instructions and analyzed by Western Blotting. The antibodies used for the western blot were anti-VAMP2 1:1000 (Synaptic Systems #104211) in 5% Milk, with Dako goat-anti-mouse 1:5000 as secondary, anti-PDI (Enzo life sciences #ADI-SPA-891-D) 1:100000 in EveryBlot, with Dako goat-anti-mouse 1:5000, anti-β-tubulin, 1:25000 in Quench (Abcam #Ab6046) with Dako goat-anti-rabbit 1:25000. For IRIS-1 the Capra custom made antibodies #613.610 or 614.610 were used, 1:1000 in Quench, with Abcam goat-anti-rabbit 1:5000. For IRIS-2 Capra custom made antibody #612.609 was used in 1:1000 in Quench, with Dako goat-anti-rabbit 1:5000.
Proximity ligation assay (PLA)
PLA was carried out according to the manufacturer’s instructions (NaveniFlex Cell MR Red #NC.MR.100) to detect and quantify protein-protein interactions [17]. In short, two primary antibodies bind to their target epitopes, located on a single protein or two nearby proteins. Antibodies used: VAMP2 (1:100, Synaptic Systems #104211, mouse monoclonal) or VAMP2 (1:100, Synaptic Systems #104008, rabbit monoclonal. Used depending on antibodies pair, as the two different species should be used, ex. mouse-rabbit), IRIS-1/ 2 antibodies (1:100, Capra, custom made #613.610, #612.609 respectively), SNAP-25 (Abcam, #EPR3275), Syntaxin 1 (1:100, Synaptic Systems #110011) and incubated overnight in 4 °C. Further, Navenibodies (antibodies conjugated to proprietary oligonucleotide arms), which bind to their respective primary antibodies were added for 1 h, 37 °C. If the Navenibodies are in close proximity, the attached oligos can generate a DNA circle, which via addition of polymerase amplifies (rolling circle amplification process) generating fluorescent puncta (fluorescent probes are bound to the amplified DNA). The high signal-to-noise enables the detection of separate proximity events, allowing for a resolution down to a single protein-protein interaction. The interactions were then visualized using Carl Zeiss 800 confocal microscopy, 10–20 Z-stacks taken per image, merged using Zeiss software, and quantified using ImageJ program (functions: nuclei count, analyze particles with threshold set up on negative control: one antibody used).
Quantification and statistical analysis
All experiments were performed in at least three independent repeats. All western blots show a representative of at least three independent repeats. The mean differences between groups that have been split into two independent variables were compared with 2-way ANOVA. Student’s t-test was used in case of two independent group comparisons, and 1-way ANOVA when three or more independent groups were compared. Normal distribution of data was verified with histogram and Q-Q plot, whereas the standard deviation between the groups was visually inspected with scatter plot. All statistical analyses were performed using GraphPad Prism 10, or R 4.3.3. Values are expressed as a mean ± SD. Statistical tests applied in each experiment are indicated in the figure legends. Bonferroni and Dunnett’s tests were used in case of multiple comparisons. All the experiments except brain samples immunostaining were repeated at least three times. In all figures, *p < 0.05, **p < 0.01 and ***p < 0.001.




留言 (0)