
記住我

The molecular dynamic simulation technique was used to evaluate the interactions of A1 with CXCR4 in silico. The stability of the interactions between the CXCR4 receptor binding pocket participant amino acids was further studied and identified through molecular docking studies [17]. The behaviors of the crystallographic ligand (ITD), A1, and AMD3100 in the binding pocket were fully monitored during 100 ns of MDs using GROMACS software v5.1.5 [18] employing GROMOS AMBER force field (amber99sb-ildn) [19]. The initial conformations of CXCR4 in complex with ITD, A1, and AMD3100 were obtained from the models reported in the previous study [17]. The CXCR4 and the target complex with the desired targets were solvated in a dodecahedral box of TIP3P water molecules with a minimum distance of 14 Å between the protein surface and the box walls, and periodic boundary conditions were assigned in all directions. The system net charge was neutralized by replacing water molecules with appropriate counter sodium and chloride ions. The van der Waals cutoff was considered 14 Å. The solvated systems were minimized through the steepest descent algorithm with 1000 kJ mol-1 nm-1 tolerance followed by a canonical ensemble (NVT) for 20 ps and an isothermal-isobaric ensemble (NPT) in a periodic boundary condition. The system’s temperature and pressure were maintained using the Berendsen thermostat [20] and the Parinello-Rahman barostat algorithm [21] at constant temperature and pressure of 310 K and 1 bar, respectively. The long-range electrostatic interactions were calculated using the particle mesh Ewald (PME) algorithm [22]. The LINCS algorithm [23] was applied to restrain all the bonds with an integration step of 1 fs. The whole system was subjected to 100 ns of molecular dynamic simulations at constant pressure and temperature. Further analysis was done over the coordinate files extracted from the trajectories.
Binding free energy calculationsThe binding affinities of the target compounds for CXCR4 were studied using binding free energy calculations using the g_mmpbsa tool and the MM-PBSA strategy [24] with GROMACS trajectories individually. The energy contribution of the key residues that bind the desired compounds was also computed.
Graphical representationDiscovery Studio 4.1 [25], VMD 1.9.2 [26], and PyMOL 2.3.4 software [27] were applied to all graphical representations and molecular images.
A1 synthesizeThe chemical processes and compound synthesis steps have been described in our previous study [17]. Briefly, the synthesis of (N-(3,4-dimethylphenyl)-2,2,2-trifluoroacetimidoyl chloride) (2a) involved combining Ph3P, Et3N, and TFA in a flask, followed by the addition of 3,4-dimethyl alanine in CCl4. After refluxing and stirring for five hours, the solvent was evaporated, and the residue was processed to yield trifluoroacetimidoyl chloride. Subsequently, for the synthesis of N, N''-thiocarbonylbis(N'-(3,4-dimethylphenyl)-2,2,2-trifluoroacetimid amide) A1, thiourea and sodium hydrogen carbonate were mixed in ether, and N-(3,4-dimethylphenyl)-2,2,2-trifluoroacetimidoyl chloride (2a) was added dropwise. After refluxing and stirring, the reaction was monitored using TLC, and the solvent was removed under reduced pressure. The resulting mixture was filtered, and the crude product was further purified by dissolving in n-hexane and filtering. The final compound was obtained after removing the solvent under reduced pressure [17, 28] (Supplementary data, Figure S1). Additionally, in our previous study, fluorine, hydrogen, and carbon nuclear magnetic resonance spectroscopy (19F-NMR, 1H-NMR, and 13C-NMR) as well as Fourier transform infrared spectroscopy (FT-IR) were used to characterize A1 [17].
Cell lines & cell cultureThe mouse embryonic fibroblast (MEF) cell line, which served as our control cells, and the CT-26 mouse CRC cells were purchased from the Pasteur Institute of Iran in Tehran. Cell lines were cultured in DMEM/F-12 (Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12) (Gibco 12,500,062) and 10% fetal bovine serum (FBS) (Gibco A4766801). The cells were incubated at 37 °C with 5% CO2.
Gene expression assayRT-PCR and flow cytometry were employed to explore how the CXCR4 receptor is expressed in CT-26 cells and compared with non-cancerous MEF cells.
According to the manufacturer's instructions, total RNA was extracted from CT-26 and MEF cells using an RNA extraction kit (Sinaclon EX6101, Iran). Spectrophotometry and gel agarose electrophoresis were employed to assess the purity and integrity of the extracted RNA. The optical density (OD) ratio of the purified RNA was checked at 260 and 280 nm to ensure that they fell within the optimal range of 1.8 to 2 (Supplementary data, Figure S2). A one-step cDNA synthesis kit (KPG-cDNA 50, Iran) was used for cDNA synthesis following manufacturer instructions. In a dedicated RNase/DNase-free microtube, a mixture of 5 μL of template RNA (ranging from 5 ng to 5 μg), one μL of either Oligo dT or Random hexamer primers, 14 μL of Master mix, and RNAse-free water to a total volume of 20 μL was prepared. The solution underwent a temperature program of 10 min at 25 °C, 60 min at 47 °C, and 5 min at 95 °C, facilitating the conversion of RNA into cDNA.
The RT-PCR technique was used to detect gene expression alterations. Primer design for specific RNA sequences was accomplished using PrimerExpress™ version 3.2 software and verified through the NCBI Primer-BLAST tool. Primer sequences are shown in Table 1. Before RT-PCR, cDNA samples were normalized to 50 ng/μL. The assessment of target gene expression, encompassing CXCR4, was quantified within cell cultures. The 2xqPCRBIO SyGreen Mix Lo-ROX PB20.11–05-s (PCRBiosystem, England) was employed for quantification. Actin-β served as the designated reference gene, and all reactions were executed in duplicate. The Rotor-Gene Q 2plex System (Qiagen) was utilized following the recommended protocol, involving an initial cycle of 95 °C for 2 min, followed by 40 cycles comprising denaturation at 95 °C for 5 s and annealing/extension at 60 °C for 25 s for template amplification. Additionally, a melting curve step was integrated into the final phase, involving 10 s at 95 °C and 10 s intervals at 0.2 °C enhancements spanning the temperature range from 62 to 95 °C.
Table 1 Primer sequences and other details of the primers used in this studyFlow cytometryFollowing the cell culture procedures outlined in previous sections, 500,000 MEF and CT-26 cells were mixed in 100 μL of staining buffer. This mixture added 2.5 μL of the PE-conjugated anti-mouse CXCR4-specific antibody (R&D systems FAB21651P). The cells were then incubated in the dark at 4 °C for 30 min. Post-incubation, a BD Bioscience FACScaliber flow cytometer (BD, USA) was employed to detect and analyze the percentage of CXCR4+ cells. Furthermore, the impact of the A1 compound on the percentage of CXCR4+ CT-26 cells was examined using flow cytometry. CT-26 cells were treated with a 60 μg/mL dose of A1 and 100 ng/mL of CXCL12 and incubated at 37 °C for 72 h. Subsequently, 50,000 cells were mixed with 100 μL of staining buffer. Then, 2.5 μL of PE-conjugated specific CXCR4 antibodies were added to the microtubes. The cells were incubated for 30 min in the dark at 4 °C. Then, we evaluated the percentage of CXCR4+ cells using a BD Bioscience FACScaliber flow cytometer (BD, USA).
Proliferation assayAn MTT assay kit (Sigma, 298-93-1, Germany) was employed to evaluate the impact of AMD3100 and A1 on cell proliferation. For CT-26 cells in a 96-well plate, a cell density of 5000 cells per well was established, with an optimal culture medium volume of 200 μL per well. Initially, CT-26 cells were plated with 100 μL of FBS-free DMEM medium per well in 96-well culture plates. Following overnight incubation, varying concentrations of AMD3100 (APExBIO, A2025, USA) and A1 were dissolved in 100 μL of medium and introduced to the respective wells. The optimal concentration of A1 and AMD3100 was determined to be 40 μg/mL based on examination of various concentrations. In addition, 100 ng/mL of CXCL12 as a proliferation stimulator and ligand of CXCR4 was added to each well. After 24, 48, and 72 h of treatment, 10 μL of sterile MTT solution (5 mg/mL) was added to each well and incubated for 3 h. Subsequently, the supernatant was removed, and 150 μL of dimethyl sulfoxide (DMSO) (Sigma, 67–68-5, Germany) was added to each well, followed by shaking for 20 min at 37 °C. The absorbance at 570 nm was measured using an ELISA microplate reader (BioTech, USA). Notably, A1 and AMD3100 were soluble in H2O with gentle warming.
Functional assaysFunctional assays were employed to confirm the binding of the compound to the CXCR4 receptor and to investigate the changes in the downstream pathway of the CXCL12/CXCR4 axis. The expression of CXCR4, matrix metalloproteinase-9 (MMP-9), and nuclear factor kappa B (NFκB) genes was assessed in two treatment groups, one with a 60 μg/mL concentration of A1 and AMD3100 (based on our previous study and obtained IC50 concentration) and the other with a control group, in the presence and absence of 100 ng/mL CXCL12 [17].
cAMP assayAdditionally, considering the role of the CXCL12/CXCR4 axis in reducing cAMP concentration, the cAMP assay was conducted using a competitive ELISA method in two treated groups with different concentrations of A1 and AMD3100 (10, 100, and 1000 nm/mL) using a cAMP ELISA kit (R&D Systems, KGE002B). Initially, CT-26 cells were seeded at a density of 106 × 5 cells per well and cultured for 24 h in DMEM/F-12 medium under FBS-deprived conditions. Subsequently, the cells in all groups were stimulated with 5 μm of forskolin (Sigma, F3917) for 30 min. Following the stimulation, each sample was treated with A1 and AMD3100 at concentrations of 10, 100, and 1000 nm/mL for 30 min [29]. After this stage, the cells were incubated for an additional 30 min with 150 ng/mL of recombinant CXCL12 protein (R&D systems, BLB041604), and they were then prepared for the ELISA step according to the manufacturer’s instructions.
Western blottingWestern blotting was used to measure the protein expression of pAKT as one of the most critical adaptor molecules in the CXCL12/CXCR4 signaling pathway and confirm CXCR4 inhibition by A1 compared with AMD3100. Cells (5 × 105 cells/mL) cultured in 6-well plates were treated with 5 and 10 µM AMD3100 and A1, followed by lysis in RIPA buffer supplemented with protease and phosphatase inhibitors. Protein content in the lysates was quantified using Bradford’s assay. Subsequently, proteins were separated by SDS-PAGE electrophoresis, transferred onto membranes, and immunoblotted with mouse anti-pAKT antibodies (R&D System, MAB887-SP) along with anti-actin β antibodies (R&D System, MAB8929-SP) for normalization. HRP-conjugated IgG (R&D System, HAF018) was utilized for membrane labeling, and protein bands were visualized through X-ray film exposure. The density and size of the bands were analyzed using Image-J software (version 1.41o, Java 1.6.0_10, Wayne Rasband, US National Institutes of Health).
Migration assayMigration/Chemotaxis Assay Kit (24-well, 8 µm) (ab235694) was used for migration assay. Initially, CT-26 cells were seeded at a density of 106 × 5 cells per well and cultured for 24 h in DMEM/F-12 medium under FBS-deprived conditions. Subsequently, 200,000 cells for each counting chamber were treated with 10 μm/mL of AMD3100 and A1 and incubated for 24 h. It is worth mentioning that CXCL12, with a concentration of 300 ng/mL and 600 μL of serum-free DMEM/F-12 medium, was added to the respective lower chambers on the day of the experiment. After a 24 h incubation in a CO2 incubator, non-migrated cells were removed from the upper compartments using a swab, and migrated cells were stained with 1.0% crystal violet for 30 min at 37 °C and then washed twice with PBS. Finally, after counting the migrated cells in 10 independent microscopic fields, a comparative diagram of the percentage of migrated cells was plotted, and images were captured from the cellular groups.
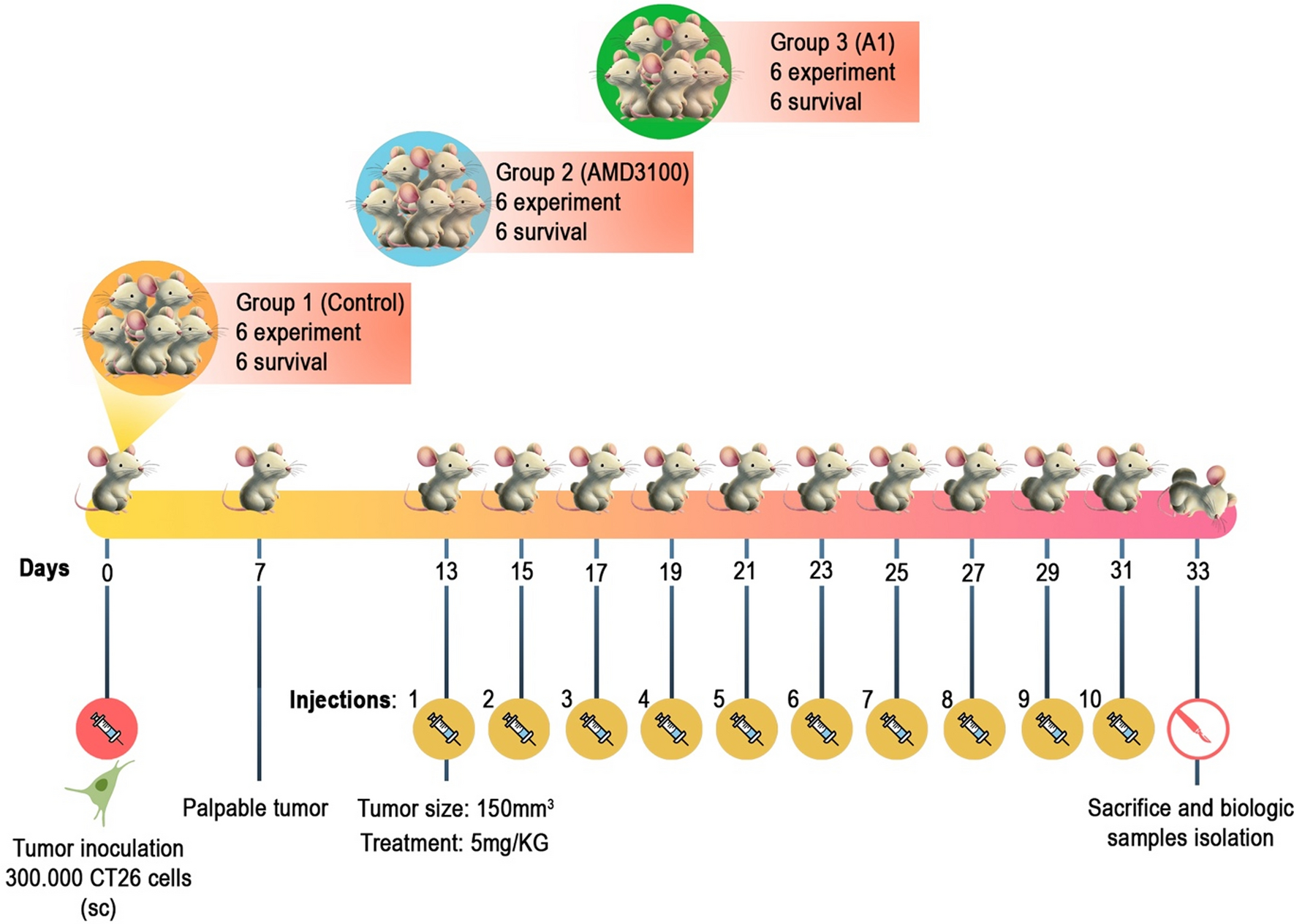
Animal modelThis study utilized 36 female BALB/c mice aged 6 to 8 weeks, weighing 15 to 17 g. They were divided into three groups of 12 each and maintained in pathogen-free conditions with proper environmental controls. The Ethics Committee of Iran University of Medical Sciences (IR.IUMS.FMD.REC.1400.590) approved the experimental procedures. The mice were allocated into three groups: Group 1 received PBS (negative control), Group 2 received AMD3100 (drug control), and Group 3 received A1 (case group). Six mice from each group were chosen for further experimentation, while six were monitored for survival analysis. Tumor inoculation was done by injecting 300,000 CT-26 cells subcutaneously into the right flank. Tumor growth was observed, and treatment began on day 13 when tumors reached 150 mm3. A 5 mg/kg dose for A1 and AMD3100 was administered intraperitoneally every other day from days 13 to 31 [30,31,32]. The control group received PBS injections (Fig. 1).
Fig. 1
An overview of the tumor inoculation and animal treatment protocol. On Day 0, mice were inoculated with tumor cells, subsequently undergoing a treatment regimen over a specified duration. Significant experimental phases are delineated, encompassing inoculation, drug administration, and sample collection
Evaluation of tregs infiltrationFollowing the removal of the tumor tissues, a scalpel was used to cut the tissues into smaller pieces for tumor cell dissociation. These pieces were then put in Falcon tubes with a cocktail of DMEM, type IV collagenase enzyme (0.2% concentration) (BIOIDEA, BI-1603, Iran), and DNAse type I enzyme (10 units/mL) (Yekta Tajhiz, YT9058, Iran). The tube was incubated for 40 min at 37 °C in a shaker incubator, with regular checks and mixing every 10 min. After the enzymatic digestion, we added a DMEM culture medium containing 10% FBS to block the enzymatic activity. The cell suspension was centrifuged at room temperature for 5 min at 1500 × g, and the enzyme-containing supernatant was discarded. The cell suspensions were then passed through a 70 μm cell strainer (SPL, Korea) to remove the possible cell clumps. After centrifuging and washing with PBS, cells were counted and prepared for flow cytometry analysis. For identifying Tregs, fluorophore-conjugated antibodies, including anti-mouse-CD4 (FITC) (100,405), anti-mouse-CD25 (PerCP) (B369413), anti-mouse-FOXP3 (PE) (B383980), and anti-mouse-CD3 (APC) (100,235) (BioLegend, USA) were used for flow cytometry analysis. Supplementary Fig. 3 shows the gating strategy.
Evaluation of CXCR4, VEGF, FGF, IL-10, TGF-β expression at mRNA & protein levelsFollowing RNA extraction from the isolated tumor tissues, preserved in RNA Later (KPG, Iran), cDNA synthesis was carried out to assess the expression of target genes, including VEGF, FGF, CXCR4, IL-10, and TGF-β, with actin-β serving as the reference gene by the RT-PCR technique. Tissue concentrations of IL-10 and TGF-β were measured using KPG ELISA kits (KPG, Iran) according to the manufacturer's instructions.
For the immunohistochemistry (IHC) staining of VEGF, we followed the protocol described by Kouvaras et al. [33]. The paraffins were removed, and the tissue sections were rehydrated in water. Natural peroxidase activity was inhibited using a 10% hydrogen peroxide solution for 10 min. Antigen retrieval involved heating the sections at 95 °C in a sodium citrate buffer (10 mM, pH 6) for 30 min. After washing with PBS, the slides were incubated with anti-VEGF primary antibody (R&D Systems, AF767-SP) at 25 °C for 50 min. Following another PBS wash, the secondary antibody was applied at 25 °C for 45 min. Lastly, the sections were counterstained with hematoxylin, covered with diaminobenzidine (DAB)(Thermo Scientific™, 34,002) color-developing solution, and evaluated under a light microscope by an expert pathologist.
Toxicity assayBiochemical and histopathological techniques were used to assess the hepatotoxicity of A1 compared to AMD3100 in the treated animals. Serum levels of liver enzymes, specifically alanine transaminase (ALT) (Delta DP, DDP01154S, Iran) and aspartate transaminase (AST) (Delta DP, DDP01159S, Iran), were measured using a Hitachi-912 autoanalyzer (Hitachi, Mannheim, Germany). Additionally, hematoxylin and eosin (H&E) staining was used to examine the alterations and morphological appearance of the hepatocytes. An expert pathologist screened the slides and subsequently interpreted the resulting microscopic observations.
留言 (0)