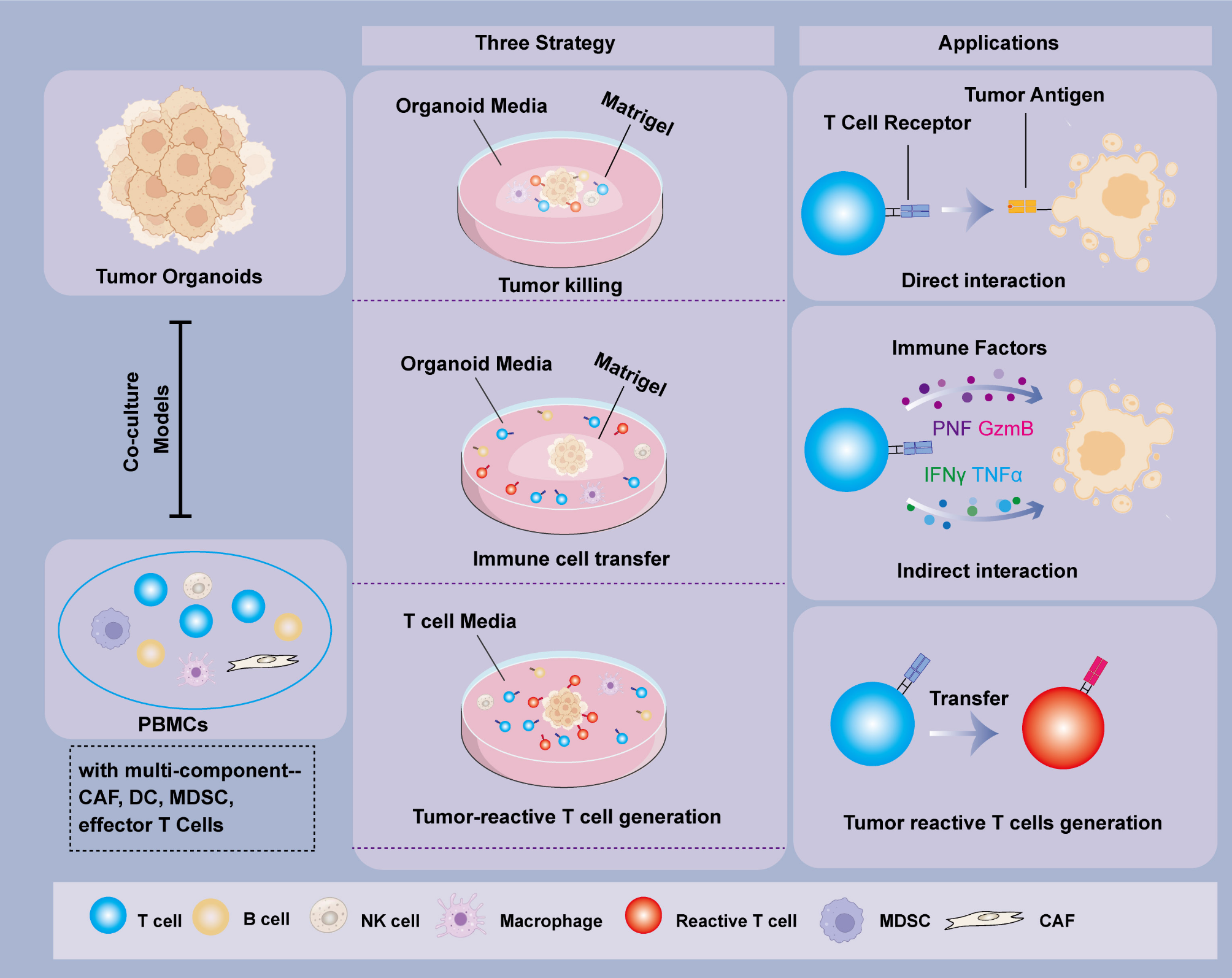
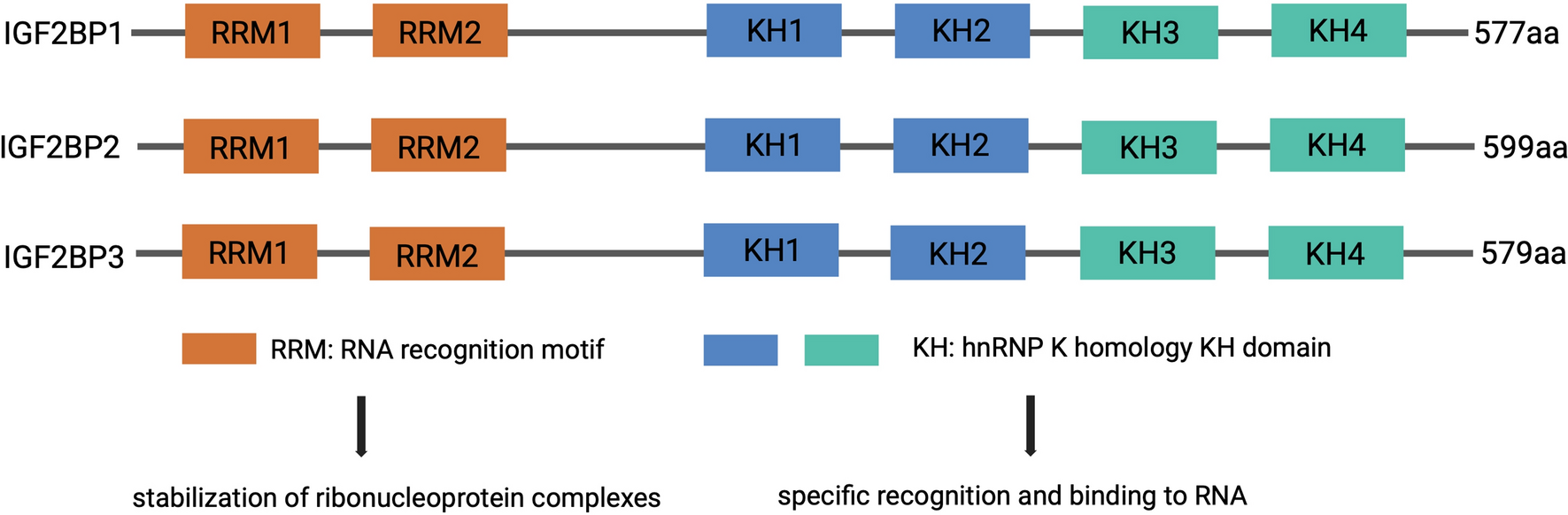
Sources and structures of tumor organoids
Long-term organoid culture models have been established from primary cancer biopsy samples [26, 27, 31]. These organoid models exhibit different success rates [32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51] (Supplemental Table 1). Tumors with high malignancy have a higher success rate, whereas those with low malignancy display a lower success rate [31, 34, 52]. Additionally, different pathological classifications are essential influencing factors. For example, the efficiency of establishing continuously propagated organoid lines from pancreatic neuroendocrine neoplasm samples was only 10%; however, that of pancreatic ductal adenocarcinoma (PDAC) samples reached 80% [31]. This affects the efficiency of constructing organoid models and limits the application scenarios of the co-culture models. Therefore, more comprehensive cultivation conditions should be explored to improve the success rate of different types of organoids, making co-culture models more widely applicable to various cancers.
Moreover, different types of organoid display different 3D structures and are primarily divided into solid, vacuolar, and mixed [31]. Compared with solid and mixed structures, vacuolar structures need a shorter digestion time, display a larger diameter, and are more susceptible to infiltration by immune cells. To better evaluate the therapeutic effect and reduce the impact of spatial structure differences, more evaluation indicators need to be considered, such as organoid diameter, the structural damage degree, apoptotic cell count, and cytokine secretion levels. Finally, due to the 3D structure, cell counting can be extremely challenging. Therefore, it is necessary to digest organoids into single cells to obtain accurate cell counts prior to co-culture.
Sources and quality of PBMCs
The PBMC sources can be divided into autologous and allogeneic sources. Several studies have demonstrated that autologous PBMCs can effectively simulate the in vitro immune response of individuals in co-cultures of pancreatic cancer and colorectal cancer [16]. However, autologous PBMC amplification and acquisition are challenging, especially in patients undergoing radiotherapy and chemotherapy. A potential solution is to obtain T-cells from different healthy individuals and test their responses to immune drugs to determine the immunogenicity of individuals.
Allogeneic PBMCs are easily obtained and can be conveniently expanded to sufficient numbers to study the sensitivity of tumor cells to immunotherapy at different time points [6]. However, the mismatch of HLA between immune and tumor cells challenges the quality of immunotherapy evaluation. HLA mismatch can lead to the inhibition of CD8 + cell activation, delays the release of tumor killer factors, and limit T-cell recognition and killing function against tumor cells, resulting in a limited immune response [53, 54].
Some scholars currently believe that using next-generation sequencing technology to match allogeneic PBMCs with tumors may be a solution [55]. Research has found that tumor organoids effectively retain most of the HLA alleles and maintain patient-specific HLA neo-antigen features. By comparing the alleles of HLA-A/- B/- C alleles on organoids with PBMCs in healthy individuals, they found that half of the allele matches were significantly attacked by T-cells [55], confirming that tumor organoids serve as killing targets and providing insights into the effectiveness and feasibility of immunotherapy.
Meng Q et al. [15] found that incubation of engineered T-cells with autologous pancreatic cancer organoids induces IFN-γ secretion within 24 h and increases by 48 h. However, incubation with allogenic organoids does not induce detectable IFN-γ secretion during the first 24 h, but IFN-γ secretion can be detected by 48 h. The delayed response to allogenic organoids was likely due to an HLA-mismatch-mediated response. This indicates that in the co-culture system, allogeneic immune cells can still recognize tumor cells and exert killing effects through other pathways. Overall, in the co-culture system of organoids and allogeneic PBMCs, the matching of MHC-1 is an important challenge that cannot be ignored and significantly affects the quality of experimental evaluation.
In addition, the quality evaluation of PBMCs also includes the absolute number and proportion of effector cells [14, 15]. CD4+ T-cells kill tumor cells through a FasL-mediated mechanism, whereas CD8+ T-cells kill tumor cells through the direct and indirect secretion of IFN-γ and granzyme B [56](Fig. 1). Therefore, the proportion and number of effector cells in PBMCs, particularly CD4+ and CD8+ T-cells, are the key factors affecting the tumor killing effect.
Proliferation and activity of PBMCs
The number of effector T-cells may be insufficient under natural conditions, leading to an insufficient killing effect on tumors [14]. Therefore, it is necessary to stimulate the immune cells to expand or express specific tumor antigens in vitro to enhance the cytotoxic effect of the immune cells. Three methods are applied to activate PBMCs (Fig. 1): (i) Activation of T-cells by exogenous compound or cytokines. For example, phorbol myristate acetate (PMA), ionomycin, and IL-2 can effectively stimulate T-cell proliferation, activation, and increase the living cell counts of CD4+, CD8+, and CD56+ natural killer cells in a co-culture or PBMC culture alone [14, 57, 58]; (ii) Simulated antigen presentation. Antibodies and synthetic small-molecule agonists are usually used, which simulate the interaction between antigen presenting cells and T-cells [14]. For example, by binding anti-CD3 and anti-CD28 antibodies with immunomagnetic beads, the magnetic beads can provide primary and co-stimulatory signals for T-cell activation and expansion [14]. CD3 is composed of multiple subunits that bind to T-cell receptors (TCRs) and transmit signals to activate T-cells [59]. Also, CD28 is a co-stimulatory molecule that binds to B7 molecules on antigen presenting cells, providing a second signal to activate T-cells. The activation of CD28 can promote the proliferation, survival, and functional enhancement of T cells, while also contributing to the formation of immune memory. (iii) Co-culture to generate specific T-cells. When co-culturing autologous PBMCs with tumor organoids, PBMCs interact with tumor cells, changing the immune cell number and type in PBMCs, even TCR genes, thus producing effector T-cells with patient tumor specificity [15, 16]. These types of T-cells express patient-specific antigens and are relatively easy to generate in sufficient quantities.
The effector (E) to target (T) cell ratio
The effector (PBMC)-to-target (Tumor organoids) cells ratio is also an unavoidable issue, related to co-culture time, conditions, cell types, and experimental purposes. Generally, the number of PBMCs needed is calculated with the formula: PBMCs needed = (V × 22,000) * n, where V denotes the volume of the dome and n represents the sample size [13]. However, studies employ different ratios depending on their specific objectives. For evaluating cell toxicity, a ratio of at least 1:1 is recommended to maximize the observation of the killing effect, thereby enabling swift and precise assessments of PBMC toxicity. For research focusing on cell interactions or signaling pathways, a ratio lower than 1:1 is preferable, as it more accurately mimics natural cellular interactions, thus revealing the subtleties of cell communication. For instance, in a study where pancreatic cancer organoids were co-cultured with autologous PBMCs to produce tumor-reactive T cells, a 1:1 effector-to-target ratio was used for co-culturing in T-cell medium for two weeks [15]. Subsequently, tumor-reactive T-cells and organoids were co-cultured at a 1:1 ratio in Matrigel for 20 h to assess the T-cells’ killing effect, and a 1:10 ratio was used for 72-hour co-cultures to evaluate cytokine release levels into the supernatant [15]. In another experiment, co-culturing gallbladder cancer with allogeneic PBMCs, the authors employed an effector-to-target ratio of 20–30:1 to amplify the attack on target cells [14].
In experiments where effector cells, such as CD4 + and CD8 + T cells, are isolated from PBMCs, their enhanced infiltration capability allows for a reduced ratio in killing experiments. In an analysis of the tumor-extrinsic toxicity of T cells in donor-matched intestinal cancer organoids, a 1:2.5 effector-to-target ratio was utilized, focusing solely on the primary effectors (CD4 + and CD8 + T cells) [13]. These ratio adjustments demonstrate the flexibility in experimental design, aiming to replicate specific biological scenarios according to research goals, thereby improving the accuracy of the outcomes.
Matrigel concentration
The extracellular matrix (ECM), the main components of Matrigel, primarily derived from non-human sources, contains tumor-related factors that can modulate the immune system and reduce anti-tumor responses, potentially causing unwanted immune reactions [60]. The ECM’s physical properties, such as its stiffness and pore size, can affect immune cell migration, direction, and invasiveness. A dense ECM can nearly block immune cell penetration into tumor organoids [16]. In certain co-culture setups, organoids are separated from the ECM using enzymes and then cultured with immune cells in suspension or on a Matrigel-free surface [61]. However, without ECM, organoids lose their 3D structure and wall-adherent growth, which can limit proliferation and alter cellular behavior [16]. Thus, fine-tuning the ECM concentration is essential for optimal organoid culture conditions.
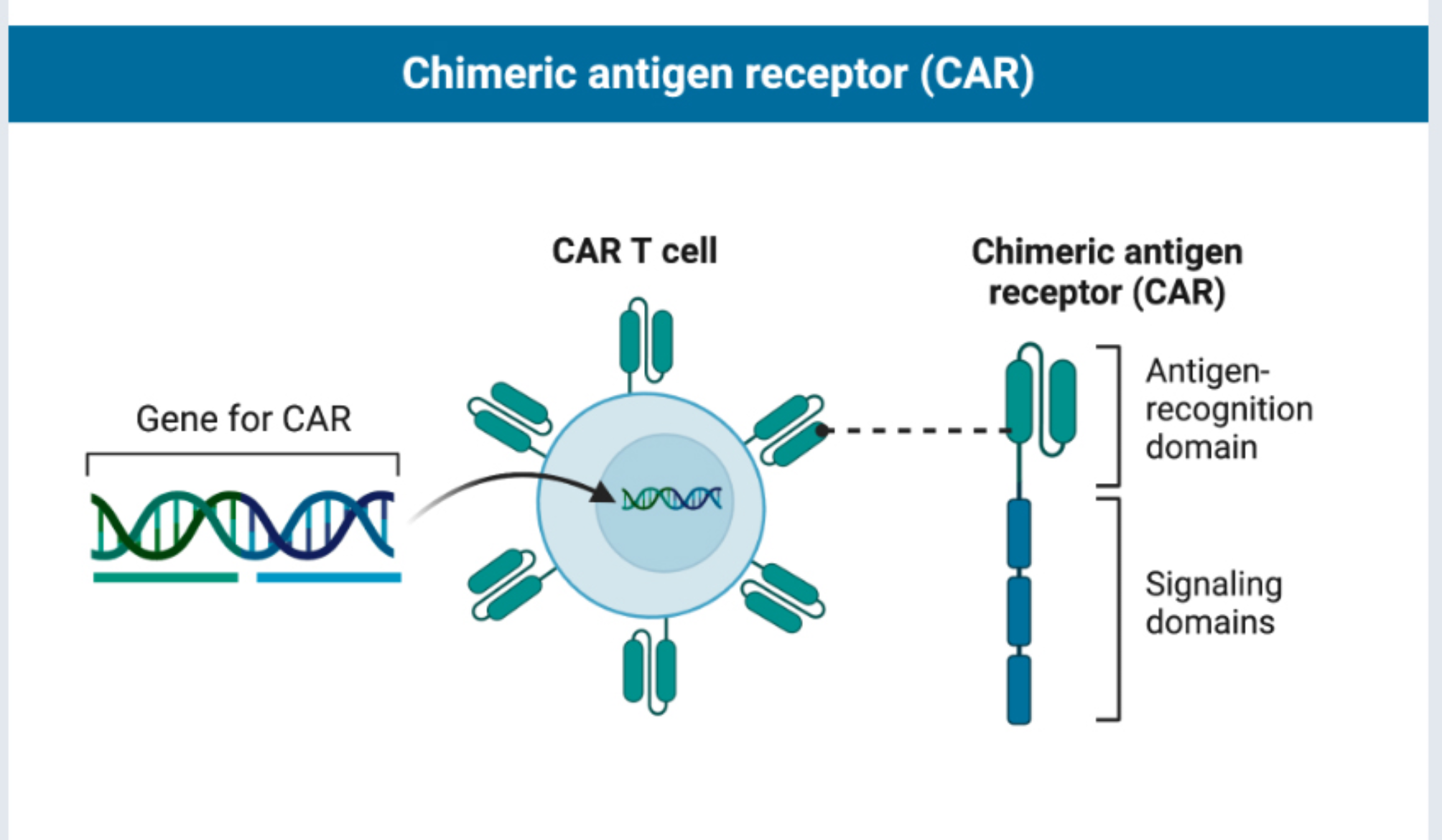
A study delving into the co-culture of cholangiocarcinoma (CCA) organoids with PBMCs examined the efficacy of diverse Matrigel concentrations [14]. The investigators discovered that a 10% Matrigel suspension adeptly maintains the organoids’ 3D structure, offering a stable scaffold without altering their shape or size. In a subsequent study validating the cytotoxic effects of CAR-T cells on bladder and kidney cancer organoids, the organoids were typically embedded in a 50% matrigel, followed by the addition of X-VIVO 15 medium containing CAR-T cells around the Matrigel dome for co-culture. This concentration of Matrigel effectively replicated the interaction between immune cells and tumor cells, establishing a reliable experimental framework for the further assessment of CAR-T cells’ therapeutic potential [62, 63]. In another experiment, researchers directly co-cultured pancreatic cancer organoids with PBMCs in T cell medium to stimulate tumor-reactive T cells. The main advantage is simplification of the experimental, which avoids non-specific immune responses from Matrigel or other 3D scaffolds and enhances immune cells’ direct exposure to tumor antigens, leading to more efficient tumor-reactive T cell generation. This approach is especially advantageous for high-throughput screening or rapid production of tumor-reactive T cells. It’s important to note that the type of tumor cells influences the suitable Matrigel concentration. For instance, gastric cancer organoids can break down Matrigel, requiring an increased concentration or more frequent medium changes [64]. In summary, the Matrigel concentration should be adjusted flexibly according to the specific requirements of each experiment.
Due to the limitations inherent in matrigel for experimental use, such as high cost, unclear origin, ethical concerns, and batch-to-batch variability, there is a growing trend towards exploring alternative matrices, such as GelMA and other hydrogels, which offer distinct advantages and are gaining traction in research settings [65]. Synthetic hydrogels, produced in various ways, are expected to replace natural Matrigel due to their higher purity and controllability. This enhances standardization and quantification, reducing non-immune-specific interference in co-culture experiments. In a prior study, intestinal organoids derived from patients were co-cultured with PBMCs and embedded in a 100% 3D hydrogel—a blend of collagen I and matrigel—for culture. These constructs were then utilized for the histological basis of multiplex immunofluorescence imaging [13]. Such a robust ECM effectively reproduces the mechanical properties of intestinal tissue and replicates key in vitro immune processes, including bystander signaling, immune cell migration, and immune cell infiltration.
Therefore, researchers are encouraged to perform gradient experiments to identify optimal concentration or explore Matrigel substitutes. The aim is to keep organoids suspended in liquid, avoiding wall adhesion, so that a thin film allows immune cells to penetrate and interact with tumor organoids.
Medium components
The exact medium components are customized for specific tumor tissues [66, 67]. To better maintain the similarity between the organoid and original tumor phenotype, more cytokines are added into the organoid culture media to ensure that tumor cells maintain a 3D structure in vitro and express the characteristics of the original tumor cells [8]. For example, WNT3A and R-spondin activate the Wnt signaling pathway, whereas Noggin and A-8301 are involved in tumor growth factor-beta inhibition. Epidermal growth factor (EGF) participates in activating the EGF signaling pathway. Forskolin is an adenylate activator, and XMU-MP-1 is a protein kinase MST1/2-inhibitor that regulates the Hippo pathway [31]. These medium components are essential for tumor organoid growth.
These cytokines can ensure better survival and proliferation of organoids in vitro; however, they can also affect PBMCs. Among them, forskolin and niacinamide inhibit T-cells, whereas IL-2 can promote T-cell proliferation [14]. Adding exogenous growth factors and small molecules during organoid growth may lead to unnecessary cloning selection, and medium components can interact with the tested drugs, resulting in an ambiguous conclusion. Therefore, a culture medium with fewer growth factors should be used to minimize clonal screening and avoid inaccurate drug treatment results. Furthermore, some co-culture systems only select a T-cells medium [14,15,16]. This method can produce tumor-reactive T-cells with organoid specificity; however, this condition cannot support the survival and proliferation of tumor cells. Therefore, further exploration is needed to determine more suitable culture medium components for co-culture to support tumor and immune cell survival and function.


留言 (0)