
記住我
The novel coronavirus, SARS-CoV-2, emerged in 2019, leading to a global pandemic with profound public health implications (1). By November 2022, there were over 628 million confirmed cases and 8 million deaths worldwide. As a betacoronavirus, SARS-CoV-2 affects both the upper and lower respiratory tracts, though severe cases often involve the lower respiratory tract, particularly in the elderly and individuals with pre-existing conditions. The tropism of the virus can vary depending on the viral strain or variant (2, 3). In response to this global crisis, vaccines such as SPIKEVAX (Moderna-NIAID) and COMIRNATY (Pfizer-BioNTech) were rapidly developed and deployed, proving instrumental in mitigating the spread of the virus and reducing disease severity (4–7). However, the emergence of new variants of concern (VOCs), including the Delta variant identified in 2020, posed significant challenges to the efficacy of these vaccines (8, 9). Variants like Delta, which exhibited higher transmissibility and partial immune evasion, marked key phases in the pandemic, though newer variants have now taken precedence. The subsequent emergence of the Omicron variant (B.1.1.529) in late 2021 (10), with over 30 mutations in the spike protein and even greater resistance to neutralizing antibodies (10–15), further underscored the need for vaccine innovations that could address these evolving threats.
The spike (S) protein of SARS-CoV-2, essential for viral entry into host cells (16), is a trimeric class I fusion protein that exists in a metastable prefusion conformation (17–19). This conformation is crucial for effective antibody-mediated neutralization (20), making the S protein a primary target for vaccine development. However, the prefusion state is inherently unstable, posing a significant challenge in vaccine design. To overcome this, the initial mRNA vaccines utilized the S2P mutation, which introduced two proline substitutions (KV986PP) that effectively stabilized the S protein in its prefusion form (19, 21, 22). Both Moderna-NIAID and Pfizer-BioNTech vaccines employed this S2P-stabilized spike protein, contributing to their success in early vaccine rollouts (23–26). Researchers have advanced this foundational work by developing a superior stabilization method involving the S6P mutation. This includes the original KV986PP mutation, along with four additional proline substitutions at positions F817P, A892P, A899P, and A942P, which further stabilize the spike protein in its prefusion form (19, 27). This design not only further stabilizes the prefusion conformation but also enhances the expression and thermal stability of the spike protein, leading to more robust immunogenicity (21, 28, 29). Comparative analyses of AlphaFold structural models of wild-type (WT), Delta, and BA.1 spike proteins, with and without stabilizing mutations, suggest that the Delta and BA.1 variants can incorporate the S6P mutations, which enhance prefusion stability and expression levels similarly to the WT spike protein. This makes the S6P-modified proteins more promising candidates for next-generation vaccines (see Supplementary Figure 1). In this study, we present RV-1730, an mRNA-based vaccine encoding the Delta variant spike protein incorporating the S6P mutation, delivered via lipid nanoparticles (LNPs). Additionally, we introduce RV-1731, a bivalent mRNA vaccine designed to target both the Delta and BA.1 variants. The primary focus of this research is the evaluation of RV-1730 as a monovalent mRNA vaccine targeting the Delta variant, assessing its immunogenicity and protective efficacy. RV-1731 is subsequently examined for its potential broader applicability, particularly in addressing emerging variants such as XBB1.5 and JN.1.These vaccines were designed not only to enhance stability and immunogenicity through the S6P mutation but also to incorporate additional modifications, such as the R682S and R685G mutations, to prevent furin cleavage, and variant-specific mutations like T19R, L452R, and D614G to further optimize antigen expression. Beyond their primary use, the efficacy of these vaccines as heterologous boosters—following initial vaccination with commercial vaccines like BNT162b2 (Pfizer-BioNTech) and mRNA-1273 (Moderna-NIAID)—was evaluated. This approach was particularly relevant given the ongoing need to improve immune responses against evolving variants. Our findings indicate that RV-1730, when used as a heterologous booster, significantly enhances neutralizing antibody titers against multiple SARS-CoV-2 variants, including Delta and Omicron. Furthermore, the bivalent RV-1731 demonstrated broad neutralizing activity and robust T cell responses, positioning it as a strong candidate for booster immunization strategies. Overall, the S6P mutation within the Delta variant spike protein provides a critical enhancement in the stability and efficacy of mRNA-based COVID-19 vaccines. RV-1730 and RV-1731 not only offer superior protection compared to S2P-based vaccines but also serve as potent boosters that can effectively combat the challenges posed by new and emerging variants. These findings highlight the importance of continued innovation in vaccine design to ensure broad-spectrum immunity against SARS-CoV-2.
Materials and methodsVaccinesAll reference vaccines, including mRNA-1273 (monovalent and bivalent with original and Omicron BA.4/BA.5) and BNT162b2 (monovalent and bivalent with original and Omicron BA.4/BA.5), were purchased from RefDrug, Inc. The RV-1730 monovalent and RV-1731 bivalent vaccines were constructed and formulated by RNAimmune, Inc. RV-1770 and RV-1771 are mRNA vaccines developed by RNAimmune to specifically target the Delta and Omicron BA.1 SARS-CoV-2 variants, respectively. Both vaccines include the S2P mutations (two proline substitutions, KV986PP) to stabilize the spike protein in its prefusion form, a feature also present in commercial mRNA vaccines such asModerna-NIAID’s and Pfizer-BioNTech’s. Additionally, RV-1770 and RV-1771 incorporate S6P mutations (four extra proline substitutions at F817P, A892P, A899P, and A942P), providing enhanced structural stability to the spike protein for Delta and Omicron BA.1, respectively.
Cell cultureHEK293T cells were purchased from American Type Culture Collection (ATCC, Manassas, VA). HEK293T cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum (FBS).
AnimalsSpecific-pathogen-free (SPF) Balb/c mice and female K18-hACE2 transgenic mice (B6.Cg-Tg(K18-ACE2)2Prlmn/J), aged 6 to 8 weeks, were obtained from Jackson Laboratories (Bar Harbor, ME) and used in this study. All mouse experiments were conducted in AIC Biotech (Rockville, MD), Denovo Biotechnology (Ijamsville, MD), and RNAimmune (Germantown, MD) in accordance with the regulations of the Institutional Animal Care and Use Committee (IACUC).
BiosafetyAll experiments with infectious SARS-CoV-2 were conducted under biosafety level 3 (BSL3) or animal BSL3 (ABSL3) at The George Mason University using standard operating procedures and were approved by the BSL3 Advisory Group, the Institutional Biosafety Committee (IBC), and the Institutional Animal Care and Use Committee (IACUC).
Animal experimentsImmunization studiesAll animal procedures were conducted by AIC Biotech (Rockville, MD, USA), Denovo Biotechnology (Ijamsville, MD, USA), and RNAimmune (Germantown, MD, USA) in compliance with the guidelines set by the Institutional Animal Care and Use Committee (IACUC). Naïve 6- to 8-week-old female Balb/c mice (The Jackson Laboratory) were randomly divided into groups of five – eight mice and vaccinated via intramuscular injection with RV-1730 (1, 5, 10 and 20 μg per mouse in 50 μl). All mice were immunized twice at 21-day intervals. Blood was taken before each immunization at 14 and 35 days after the first immunization via submandibular bleeding. The blood was allowed to clot overnight at 4°C before the serum was harvested by centrifugation at 10,000 xg for 10 minutes at 4°C. Samples were stored at 4°C until further analysis. For homologous and heterologous as well as booster administration, female Balb/c mice (The Jackson Laboratory) received intramuscular injections of 5 µg each of BNT162b2, mRNA-1273, and RV-1730 on days 0 and 21 for as a prime-boost vaccination. To standardize dosing across different vaccine sources, mRNA concentration measurements were conducted for BNT162b2, mRNA-1273, and RV-1730. This ensured comparable dose levels, with adjustments made for any differences in mRNA content to align with the commercial vaccines. Then, a booster dose of 2.5 µg for each vaccine was administered on day 72 post the first immunization. Blood samples were collected at 14, 35, 49, 63, 77, and 105 days after the initial immunization. Spleens were harvested at day 105 for T cell immunity experiments.
Live virus challengesFemale K18-hACE2 mice (B6.Cg-Tg(K18-ACE2) Primn/J, The Jackson Laboratory), aged 6-8 weeks, underwent immunization on day 0 and day 14 at Nobel Life Science (NLS) in Sykesville, MD, USA in compliance with the guidelines set by the IACUC. On day 21, the mice were transported to George Mason University (GMU; Manassas, VA, USA) and acclimated for 7 days in ABSL-2 containment. Blood samples were collected on day 27 at GMU to prepare serum before virus challenge. Serum aliquots from day 13 and day 27 were sent to the sponsor for further analysis. On day 28, the mice were transferred to ABSL-3 containment and intranasally challenged with either 5.5 x 104 PFU/mouse of SARS-CoV-2/human/ITA/INMI1/2020 (wild type) or 1.5 x 104 PFU/mouse of SARS-CoV-2 B.1.1617.2 Delta variant. Daily monitoring for mortality, body weight, body temperature, and signs of distress was conducted for 13 days (wild type) or 14 days (Delta) post-challenge. Surviving animals were euthanized 13-14 days after the challenge.
In vitro transcription (IVT)The optimal conditions for in vitro transcription (IVT) were determined, and the IVT process was carried out on the mRNA-1730 producing plasmid. Initially, the plasmid was linearized using XhoI and purified. The mRNA was then synthesized through the following steps: RNase-free water and NTPs were added to a reaction tube. CleanCap AG (TriLink) was added to the tube and vortexed for thorough mixing. The liquid was briefly spun down to collect. 10X Transcription Buffer was added, and the mixture was vortexed again. The liquid was briefly spun down. DNA template was added to the reaction. Enzymes were added to the reaction. The mixture was well-mixed by flicking or inverting the tube 10 times, and the liquid was briefly spun down. The reaction was incubated at 37°C for 4 hours. The components of the 10X Transcription Buffer consisted of 400 mM Tris-HCl (pH 8), 100 mM DTT, 20 mM Spermidine, 0.02% Triton X-100, 165 mM Magnesium Acetate, and DNase/RNase-Free Water. In the final reaction, the concentrations were adjusted to 1X Transcription Buffer, 5 mM ATP, 5 mM CTP, 5 mM GTP, 5 mM N-1metyl-pseudoUTP, 4 mM CleanCap AG, Murine RNase Inhibitor (1 unit/μl), E. coli Inorganic Pyrophosphatase (0.002 units/μl), T7 RNA Polymerase (8 units/μl), and 25-50 ng/μl linear DNA.
RV-1730 and RV-1731 formulationRV-1730 and RV-1731 are lipid nanoparticles (LNPs) carrying mRNA encoding the prefusion stabilized spike protein (S6P) of SARS-CoV-2. mRNA-LNPs were generated by using the NanoAssemblr Ignite (Precision NanoSystems). The formulated LNPs were collected in a labeled tube. After motor repositioning, the lid was opened safely, and the tube labeled “mRNA-LNP” was removed for immediate characterization. For LNP characterization, 25-50 μl of the sample fraction was mixed with ultra-pure water and particle size was measured on Zeta Sizer (Malvern Pananalytical). The sample was transferred to a dialysis cassette and dialyzed overnight. The LNP formulations were concentrated using ultra-centrifugal filters, filtered through a 0.22-μm filter, and the final mRNA concentration and encapsulation efficiency were measured using the Quant-it Ribogreen Assay Kit (Invitrogen). LNPs were stored at 4°C (in PBS) or -20°C.
Western blotLysates from transfected HEK293T cells were prepared in 200 μl RIPA buffer (Thermo Scientific) with a proteinase inhibitor cocktail in a 6-well plate (one well per sample). The plate was then placed on ice for 30 minutes and gently mixed. Subsequently, the cell lysates were transferred to 1.5 ml tubes, briefly sonicated, and centrifuged for 15 minutes at maximum speed. BCA assays were employed to determine protein concentrations in the lysates. For gel electrophoresis, inactivated cell lysates (10-20 μg per lane) were heated at 95°C for 5 minutes, separated on an SDS-PAGE gel (4-12% NuPAGE, Invitrogen) with MES Running buffer for 30 minutes at 200 V, and transferred onto a PVDF membrane using the iBlot2 system (Invitrogen). Following blocking with 5% milk/TBST (1x Tris-buffered saline containing 0.1% Tween-20), the membranes were incubated with primary antibodies in 5% milk/TBST at 4°C overnight. The primary anti-S monoclonal antibodies for S1, S2, and RBD were purchased from Invitrogen (S1: MA5-36249; S2: MA5-36254; RBD: MA5-36253, respectively). After washing with PBST, secondary antibodies conjugated with HRP were applied to the membrane in 5% milk/TBST for 1 hour at room temperature. Chemiluminescence for image acquisition was achieved using Clarity Western ECL substrate (Bio-Rad), and band intensities were quantified using an Azure ChemiDoc Imaging System (Azure Biosystem).
ELISAMaxiSorp plates (BioLegend) were coated with 100 μl of recombinant S1 or RBD (1 μg/ml) in sodium bicarbonate buffer overnight at 4°C. The antigens used for coating included S1 proteins for multiple SARS-CoV-2 variants: Alpha (Sino Biological, Cat #40591-V08H12), Beta (Sino Biological, Cat #40591-V08H15), Gamma (Sino Biological, Cat #40589-V08H26), Delta (Sino Biological, Cat #40150-V08B1), BA.1 (Sino Biological, Cat #40591-V08H41), and BA.2 (Sino Biological, Cat #40589-V08H28). Additionally, RBD proteins were used for the Wild Type (Sino Biological, Cat #40592-V08H), Alpha (Sino Biological, Cat #40592-V08H82), Beta (Sino Biological, Cat #40592-V08H85), Delta (eEnzyme, Cat #SCV2-RBD-IN2P), BA.1 (Sino Biological, Cat #40592-V08H121), BA.2 (Sino Biological, Cat #40592-V08H123) and XBB.1.5 (Sino Biological Cat # 40591-V08H47).The wells were washed 3 times with PBS-T and incubated the plates with 200 μl of blocking buffer for 2 hours at room temperature (RT). After washing 3 times with PBS-T, the plate was incubated with 100 μl of sera for 2 hours at RT. And the plate washed 5 times with PBS-T and incubated with HRP-conjugated secondary antibody for 1 hour at RT. Finally, the plate was washed 5 times with PBS-T and incubated the plates with 100 μl of 1x TMB substrate for 10 minutes. The reaction was stopped by adding 100 μl of 1N-HCl and read at 450 nm using the Cytation7. For reciprocal endpoint IgG titers, 2-fold serial diluted sera were added to the wells immobilized with different spike proteins. All procedures were the same as described above. For isotyping of binding antibodies, MaxiSorp plates (BioLegend) were coated with 100 μl of recombinant S1 protein (1 μg/ml) in sodium carbonate buffer and left to incubate overnight at 4°C. The plates were then washed three times with PBS-T and blocked with 200 μl of blocking buffer for 2 hours at room temperature (RT). Following another three PBS-T washes, 100 μl of sera was added and incubated for 2 hours at RT. After five additional PBS-T washes, plates were incubated with HRP-conjugated anti-mouse IgG, IgG1, IgG2a, or IgG2c (Abcam) for 1 hour at RT. Following five more PBS-T washes, 100 μl of 1x TMB substrate was added for 10 minutes. The reaction was stopped by adding 100 μl of 1N-HCl, and optical density was read at 450 nm using the Cytation7.
Flow cytometryTo evaluate the expression of spike proteins on HEK293T cells transfected with mRNA and mRNA-LNP, flow cytometry was performed. The culture media was removed from the transfected cells by aspiration and the cells were washed with 1x PBS. The cells were detached by adding 300 μl of 0.05% Trypsin-EDTA, incubated for 5 minutes at 37°C, 5% CO2 incubator and counted in 1 ml of complete media. 2 x 105 cells were aliquoted in a 96 round bottom well plate and centrifuged for 5 minutes at 1,800 rpm to pellet down the cells. The cells were washed twice with 1x PBS and twice with FACS Cell Staining Buffer. Cells were resuspended in 100 μl of staining buffer containing primary antibody at a concentration of 5 μg/ml at 4°C for 1 hour. After the cells were washed 2 times with FACS Staining buffer, they were resuspended in 100 μl of staining buffer containing 1:200 diluted anti-Rabbit IgG(H+L)-FITC for Clone H4 and anti-FLAG M2-FITC for hACE2 (5 μg/ml) at 4°C for 30 minutes, then washed twice with FACS staining buffer. The stained cells were resuspended in 200 μl of PBS containing 100 ng/ml of DAPI (BD Pharmingen, 564907). Flow cytometry was performed using a BD FACSCelesta for data acquisition and a FlowJo V10.7.2 for data analysis.
ELISpotSpleens were harvested from mice and processed into single-cell suspensions in RPMI1640 media supplemented with 10% heat-inactivated fetal calf serum and penicillin/streptomycin (R10 media). To eliminate red blood cells, RBC lysis buffer (KD Medical) was utilized, and the cells were subsequently resuspended in R10 media to terminate the lysis process. Following cell counting, 200,000 cells per well were seeded into 6-well plates, which were then utilized for the Mouse IFN-γ ELISpot PLUS (HRP) kit (Mabtech). The cells were stimulated for 16 hours at 37°C with 2 μg of PepMix™ SARS-CoV-2 (Spike B.1.617.2/Delta) from JPT Peptide Technologies GmbH, while media alone served as a negative control. Spots were developed following the manufacturer’s instructions, and their quantification was performed using Cytation7.
Intracellular cytokine stainingThe splenocytes were counted, and 1 x 106 cells per well were seeded into 96-well plates. Subsequently, the cells were stimulated in the presence of a protein transport inhibitor (Brefeldin A, Biolegend) for 16 hours at 37°C with 2 μg of PepMix™ SARS-CoV-2 (Spike B.1.617.2/Delta) from JPT Peptide Technologies GmbH, while media alone served as a negative control. After stimulation, Splenocytes were stained as follows; Centrifuge the plate at 400 x g for 8 minutes at 4°C, then carefully resuspend the cell pellets in the remaining fluid. Wash the cells twice with azide- and serum/protein-free PBS, ensuring the supernatant is completely decanted. Add 100 µL of the Fixable Viability Dye (FVD-V450) solution (1:1000 dilution in PBS) to each well and mix immediately by pipetting. Incubate for 30 minutes at 2–8°C. Wash the cells twice with Cell Staining Buffer. Block non-specific Fc-mediated interactions by incubating the cells with 0.5 µg of anti-Mouse CD16/CD32 antibody in 50 µL of Cell Staining Buffer for 10 minutes at 2–8°C. Add 50 µL of primary antibody mixtures (anti-CD3-FITC, CD4-PerCP-Cy5.5, and CD8-BV650, Biolegend) and incubate for 30 minutes at 2–8°C. Wash cells twice with Cell Staining Buffer. After the last wash, discard the supernatant. Fix the cells by adding 200 µL of IC Fixation Buffer to each well. Incubate for 60 minutes at room temperature. Centrifuge at 400–600 x g for 5 minutes at room temperature and discard the supernatant. Add 200 µL of 1X Permeabilization Buffer to each well and centrifuge at 400–600 x g for 5 minutes at room temperature. Discard the supernatant. Resuspend the pellet in residual volume and adjust to approximately 100 µL with 1X Permeabilization Buffer. Block with 2% normal mouse/rat serum by adding 2 µL directly to the cells and incubate at room temperature for 15 minutes. Without washing, add the recommended amount of directly conjugated antibody mix (anti-IFN-γ-PE, IL-4-APC, Biolegend) for detection of intracellular antigen(s) and incubate for at least 30 minutes at room temperature. Centrifuge at 400–600 x g for 5 minutes at room temperature and discard the supernatant. Wash cells with 200 µL of 1X Permeabilization Buffer and centrifuge again. Resuspend stained cells in 200 µL of PBS. Perform flow cytometry (FACS) analysis. Ensure compensation for spillovers when multiple fluorophore-conjugated antibodies are used.
Quantification of intracellular cytokine productionThe splenocytes were counted, and 2 x 105 cells per well were seeded into 96-well plates. Subsequently, the cells were stimulated for 72 hours at 37°C with 2 μg of PepMix™ SARS-CoV-2 (Spike B.1.617.2/Delta) from JPT Peptide Technologies GmbH, while media alone served as a negative control. Following stimulation, the plates were centrifuged, and the supernatant was collected and frozen at -80°C for cytokine detection. The analysis of secreted cytokines was conducted using a murine 11-plex kit through a multiplex bead-based technology (Luminex) assay, employing a Luminex100/200 instrument (Luminex). Supernatants were subjected to a 2-fold dilution before measurements and analyses to assess the levels of various cytokines.
Pseudovirus neutralizing assaysGeneration of SARS-CoV-2-pseudovirus particlesMurine Leukemia Virus (MLV) particles pseudotyped with a SARS-CoV-2 Spike protein construct were generated in HEK293T (ATCC) cells. All the plasmid DNAs were purified with ZymoPURE II Plasmid Midiprep Kit (Zymo Research). In brief, 8 million HEK293T cells were plated into a 10-cm tissue culture dish (Santa Cruz) in 16 ml DMEM (Genesee Scientific) containing 10% FBS (Corning Life Sciences) without any antibiotics. The following day, the cells were transfected with 8 µg pTG-Luc, 6 µg pCMV-MLVgag-pol and 6 µg pcDNA3.1-SARSCoV-2-SpikeΔC19 of different variants using Lipofectamine 3000 reagent (Thermo Fisher). The cells were cultured for an additional 48 hours. The supernatant was collected into a 50-ml Falcon tube and spun at 290 ×g for 7 minutes. The supernatant (pseudotyped virus solution) was then passed through a 0.45 μm filter (Santa Cruz) using appropriate syringe. The pseudotyped virus solution was then aliquoted into cryovials and stored at -80°C. Each 10-cm cell culture dish produces about 16 ml SARS-CoV-2-PP. The SARS-CoV-2-PP was tested for the quality control with HEK293-ACE2 cell line (created at Codex BioSolutions).
Neutralizing assay with pseudovirusDue to limited serum availability, a pooled serum sample was prepared for each group and then serially diluted threefold in quadruplicate. It was heat inactivated by incubating the serum at 56°C for 30 minutes. The day before the infection, 7.5 x 103 HEK293-ACE2 cells were plated into a 384-well white clear plate (Corning Life Sciences) precoated with Poly D Lysine (Trevigen) in 15 µl culture medium (DMEM containing 10% Fetal Clone II Serum, Fisher Scientific). The cell plate was placed in a CO2 incubator at 37°C. On the 2nd day, the serum to be tested was diluted in the culture medium on a 96-well compound plate. They are 5X of the final concentrations. 65 µl of SARSCoV-2 MLV pseudoviruse particles (pp) were mixed with 26 µl of the testing sample prepared above and incubated at 37°C for 1 hour. After the medium in each well of 384-well cell plate was removed, 17.5 µl of each serum-pp mixture was added into each well. The plate was centrifuged at 54 xg for 15 minutes at 4°C and additional 7.5 µl of the culture medium was then added into each well. Luciferase activities were measured with Firefly Luciferase Assay Kit (Codex BioSolutions Inc). IC50 values were calculated based on curve fitting in GraphPad Prism (data was normalized as the percentage of infectivity).
Plaque reduction neutralization (PRNT) assayThe neutralizing titers of serum specimens collected on study day -1, day 13 and day 27 were assessed using plaque reduction neutralization test (PRNT). All sera were heat inactivated by incubation at 56°C for 30 minutes prior to testing. Six serial two-fold dilutions (1/2 to 1/64) of each serum were prepared in EMEM and 25 μl of each dilution was combined with 25 μl of SARS-CoV-2/human/ITA/INMI1/2020 viral stock, mixed and incubated at 37°C for 1 hour. Supernatant was removed from Vero-E6 cells seeded the night before in 12 well plates and replaced with 300 μl of Eagle Minimum Essential Medium (EMEM) in each well. The incubated virus + diluted serum mixtures were added on top of Vero E6 monolayer, and plates were incubated at 37°C/5% CO2 for 1 hour. Plates were shaken every 10-15 minutes during incubation. After one-hour incubation, 1.5 ml overlayer containing 2X EMEM and 0.6% agarose at a ratio of 1:1 was added to the plates. Plates were incubated for 72 hours at 37°C/5% CO2 after solidification of agarose at room temperature. Following incubation, cells were fixed by adding 0.5ml of 10% formaldehyde into each well on top of the agarose and incubating at room temperature overnight. The palettes of agarose were gently removed, and the cellular monolayers were stained for 10 to 15 minutes with the addition of 0.5 ml of 1% crystal violet. After staining, the plates were washed with water several times to remove excess stain solution and plaques were manually enumerated for each viral dilution. The neutralizing titers were calculated as the reciprocal of the lowest dilution that resulted in a greater than 50% reduction (PRNT50) or 90% reduction (PRNT90) in PFU relative to negative control sera. Human sera collected from two fully vaccinated males and serum from naïve mice were used as a positive and negative controls, respectively.
HistologyA portion of right lung tissue of each mouse euthanized was collected and placed into 10% formalin. Lung tissue samples fixed with formalin were removed from ABSL3 and shipped to Histoserv (Germantown, MD) for histopathological assays.
Statistical analysisThe data were presented as the mean ± standard deviation (SD). Statistical analyses were carried out using GraphPad Prism 10 (GraphPad Software, La Jolla, CA). Group differences were assessed using Student’s t-test, one-way ANOVA with Dunnett’s multiple comparisons test and two-way ANOVA with Tukey’s multiple comparisons test. A significance level of P < 0.05 was deemed statistically significant.
ResultsDelta S6P mRNA is expressed at a significantly higher level than S2P in the LNP systemInitially, we evaluated the expression of the SARS-CoV-2 Spike S6P antigen in HEK293T cells post-transfection with custom-designed mRNA targeting various viral variants. Within the HEK293T cell system, we investigated the binding specificity and affinity of the SARS-CoV-2 spike protein with human ACE2 (hACE2). Different forms of SARS-CoV-2 mRNAs, including S WT S2P/D614G (100% pseudouridine), Delta S2P, and Delta S6P mutation were utilized for transfection. Notably, the S6P-stabilized spike protein exhibited an elevated expression level compared to S2P in the WT and Delta variants (Figure 1A; Supplementary Figure 2). The two bands observed in WT 2P are due to the absence of furin cleavage site mutations R682S and R685G, which are present in the Delta 2P and 6P variants. The mRNA encoding the S6P-stabilized SARS-CoV-2 spike protein demonstrated the highest expression and manifested robust binding activity to hACE2 and Clone H4 when formulated with lipid mixture, designated as RV-1730 (Figures 1B–D). These findings imply that RV-1730, featuring the S6P-stabilized spike protein, holds considerable promise as a vaccine candidate against SARS-CoV-2, with the potential to induce robust immunogenic responses in vivo.
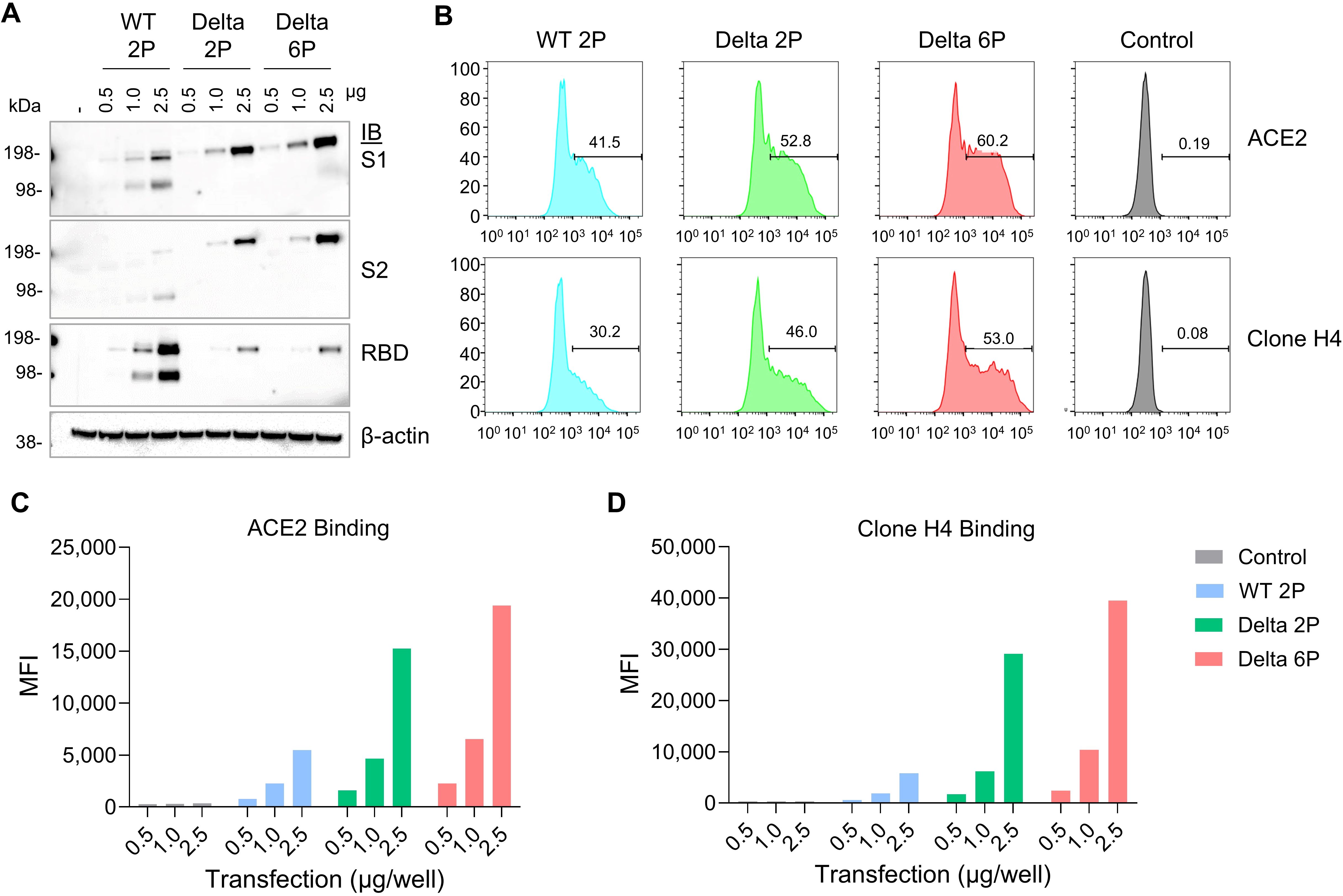
Figure 1. In vitro expression of mRNA encoding Spike protein with 2P or 6P mutations in various strains. (A) HEK293T cells were transfected with mRNA encoding different forms of the SARS-CoV-2 S protein. After 48 hours, cell lysates were analyzed by Western Blot using an anti-SARS-CoV-2 S2-specific antibody. (B–D) the cells underwent flow cytometry to measure surface expression, with staining performed using a Flag-tagged ACE2 receptor and the anti-S antibody clone H4 (B) The mean fluorescence intensity (MFI) was calculated to assess the cell surface expression levels, using the anti-Flag antibody (C) and the anti-S antibody clone H4 (D).
RV-1730 induces robust humoral responses in mice compared to S2P-based vaccinesWe then examined the immunogenicity of the RV-1730 Delta variant with S6P mutations and assessed functional immune responses, such as IgG titer and neutralizing antibody, against various SARS-CoV-2 variant spike proteins, including the WT, Alpha, Beta, Delta variants, and Omicron BA.1 and BA.2 variants. In brief, naive female Balb/c mice aged 6 to 8 weeks were intramuscularly vaccinated with RV-1730 at doses of 1, 5, 10, and 20 μg per mouse on day 0 and day 21 (Figure 2A). ELISA was used to evaluate mouse IgG titers against SARS-CoV-2 spike protein fragment S1. RV-1730 induced robust IgG titers against SARS-CoV-2 S1 for the WT, Alpha, Beta, Delta, and Omicron BA.1 and BA.2 variants (Figure 2B). The results demonstrated dose-dependent IgG titers across all variants, albeit with slight variations in intensity. To determine if the serum from immunized mice with RV-1730 could inhibit SARS-CoV-2 infection, HEK293T cells were stably transfected with ACE2 and exposed to SARS-CoV-2 pseudotyped particles (PP) from murine leukemia virus (MLV). MLV pseudovirus expressing spike proteins from pcDNA3.1-SARS-CoV-2 vectors was used for spike protein expression. The blood sample from the group exhibiting the highest IgG titer against SARS-CoV-2 spike protein S1 and RBD (e.g., RV-1730 immunized at 10 μg) 35 days post-immunization was utilized in the neutralization assay. In these assays, the serum effectively prevented infection of HEK293-ACE2 cells by very low concentrations of SARS-CoV-2 pseudotyped particles (PP) from different variants (WT, D614G, Alpha, Beta, and Delta) (Figure 2C). At 35 days post-immunization, the serum exhibited potent neutralizing activity against all tested variants, with a range of 559 – 4,762 serum IC50 in GMT (Figure 2C). The Gamma variant was the most sensitive, while the Alpha variant was the most resistant to RV-1730-immunized serum (0.021 and 0.179% of IC50, respectively) (Figure 2D). RV-1730 demonstrated strong immunogenicity against early SARS-CoV-2 strains in mice.
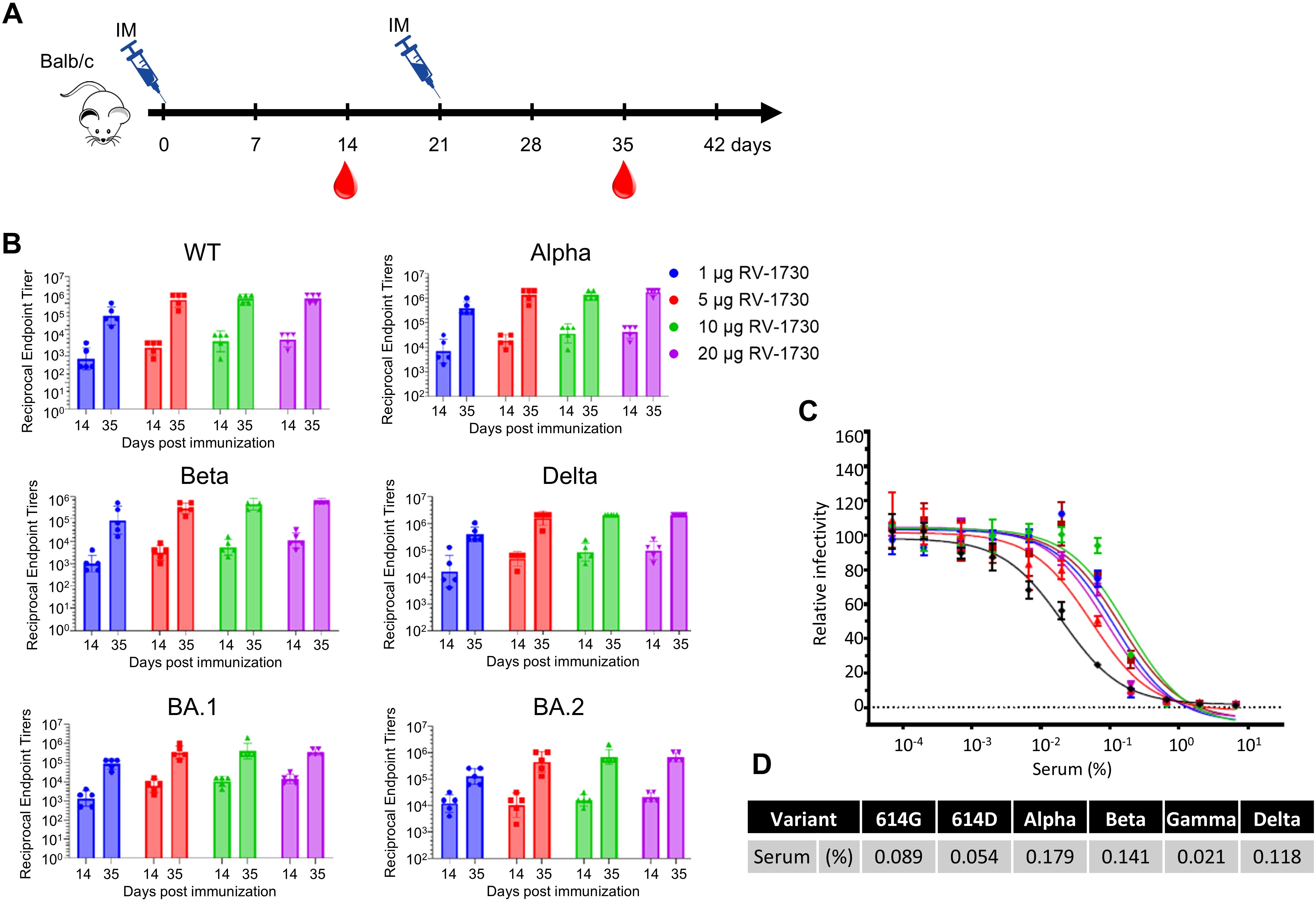
Figure 2. Endpoint IgG titers and neutralizing antibody titers of RV-1730 immunized sera against different variant spike proteins. (A) Immunization schedule of animal experiment. Female Balb/c mice were randomly divided into groups of mice (n = 5~8) and vaccinated via intramuscular injection with RV-1730. All mice were immunized twice at 21-day intervals. Blood was taken before each immunization at 14 and 35 days after the first immunization via submandibular bleeding. (B) Reciprocal endpoint titers of serum samples. Serum samples at the indicated time were tested in ELISA against S1 protein of WT, Alpha, Beta, Delta, Omicron BA.1, And BA.2. Each symbol represents a serum sample, and the bar is the geometric mean of the group. (C) Serum neutralizing titers of the serum in a pseudovirus neutralization assay. Pooled serum samples from the 10 µg group (n=5), collected on day 35, were tested using pseudovirus particles representing various variants. (D) The 50% inhibitory dilution titers (IC50) were calculated as the percentage of the diluted serum.
RV-1730 induces strong and Th1/Th2-balanced T cell responses in miceWe further evaluated T cell immune responses to RV-1730 in mice. Mice were immunized with RV-1730 at doses of 1, 5, and 10 μg per mouse, and their sera were analyzed for antibody isotypes. Across all three concentrations tested, RV-1730 immunogens generated both IgG2a/c and IgG1 subclass S-binding antibodies, indicating a balanced Th1/Th2 response (Supplementary Figure 3). This observation was further validated through a multiplex cytokine secretion assay using the Luminex 100/200 system. Upon restimulation with peptide pools (S1 and S2) corresponding to the S protein, splenocytes from RV-1730 immunized mice exhibited a well-balanced secretion of Th1 cytokines (IFN-γ, IL-2, TNF-α) and Th2 cytokines (IL-4, IL-5, IL-13) (Figure 3A), demonstrating a dose-dependent response. Quantification of SARSCoV-2 antigen-specific IFNγ-producing T cells by ELISpot revealed a robust induction of IFN-γ+ ELISpots, particularly with the Delta S1 peptide pool, while S2 peptide pools exhibited a comparatively lower frequency (Figures 3B, C). Further assessment of cytokine patterns in vaccine-induced memory T cells through intracellular cytokine staining (ICS) confirmed that the IFN-γ+ T cell response, stimulated ex vivo with Delta S1/S2 peptide pools, supported the notion that the Delta S1 peptide pool induced activation of both CD4+/IFN-γ+ and CD8+/IFN-γ+ T cells (Supplementary Figure 4). These findings collectively indicate that the RV-1730 vaccine can elicit a strong T cell immune response, characterized by T cells capable of producing a balanced Th1/Th2 cytokine profile.
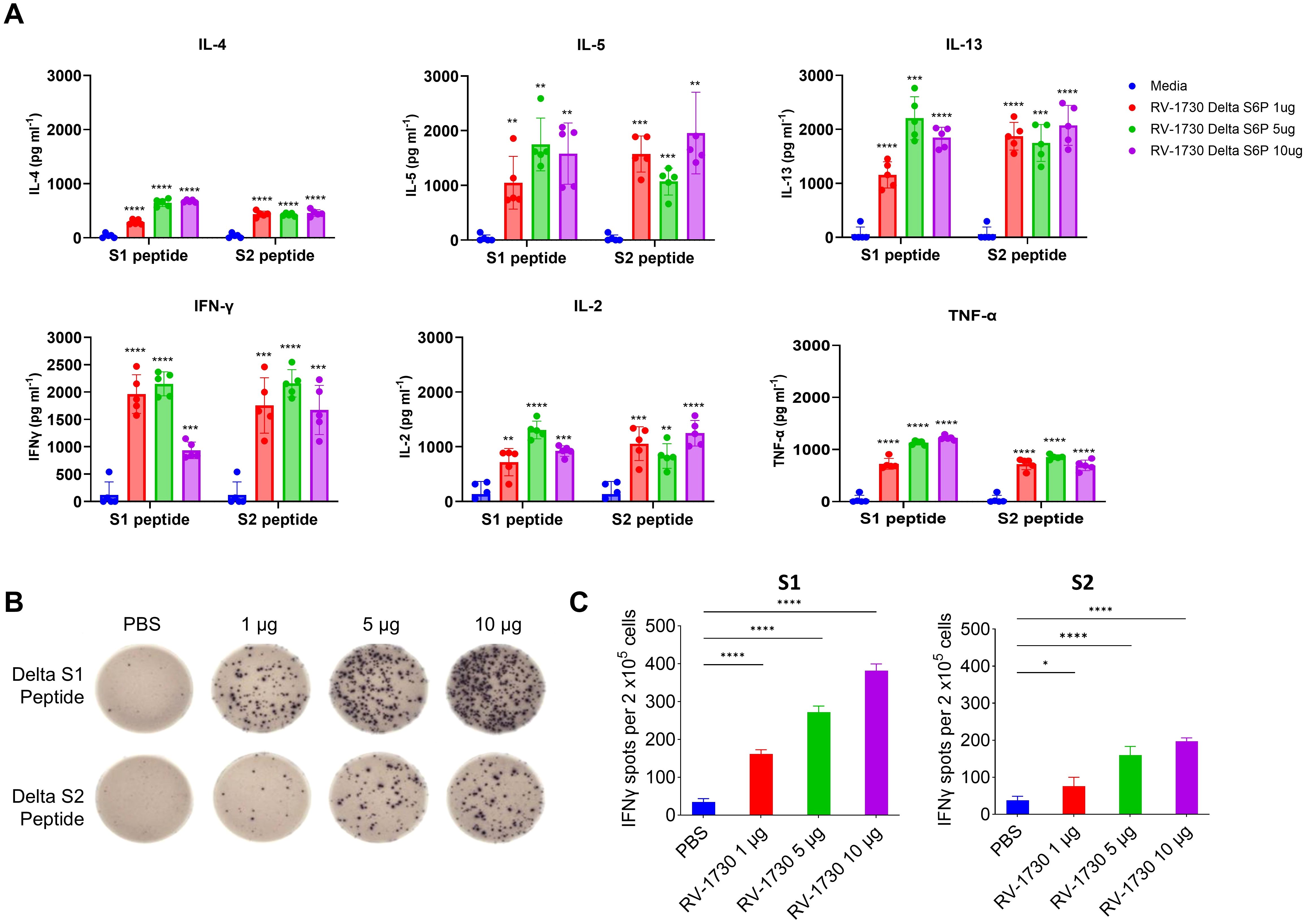
Figure 3. T cell responses in mice immunized with RV-1730. (A). 9 weeks post-boost, splenocytes were isolated from 5 mice per group and re-stimulated with no peptides or pools of overlapping peptides from SARS-CoV-2 S protein. After 72 hours, the culture supernatants were harvested by centrifugation and the secreted Th1-cytokines (IFN-γ, IL-2, TNF-α) and Th2-cytokines (IL-4, IL-5, IL-13) were measured using the bead-based, 11-plex TH1/TH2 mouse ProcartaPlex multiplex immunoassay (Thermo Fisher Scientific). Fluorescence was measured with a Luminex100/200 system and analyzed with ProcartaPlex Analyst 1.0 software (Thermo Fisher Scientific). Below the lower limit of quantification were set to zero. (B, C) IFN-γ ELISPOT analysis on splenocytes. Splenocytes (2 x105 cells per 96 well) were plated onto mouse IFN-γ ELISpot plates (Mabtech) and re-stimulated ex vivo with pools of overlapping peptides from SARS-CoV-2 Delta Spike for 16 hours. Images were taken (B) and quantified by using Cytation7 (C). *P<0.05 and ****P<0.0001 (one-way ANOVA with Dunnett’s test). **p<0.01, ***p<0.001.
Immunization of RV-1730 provides complete protection against SARS-CoV-2 ancestral WT strain challenge in miceIn a comprehensive assessment of the RV-1730 vaccine’s efficacy and dose-response, we utilized a mouse model of lethal infection by immunizing human ACE2 knocked-in K18-hACE2 mice with RV-1730. The immunizations were administered via intramuscular injection into the hind leg on study days 0 (prime) and 14 (boost). In this challenge study, we shortened the dosing interval from 21 to 14 days to expedite the study timeline and evaluate immune responses within a shorter timeframe. This adjustment was based on preliminary data indicating that a robust immune response could still be achieved with the condensed schedule. On day 28, the mice were transferred to ABSL-3 containment and intranasally challenged with 5.5 x 104 PFU/mouse SARS-CoV-2/human/ITA/INMI1/2020 (wild type). Throughout the thirteen-day post-challenge period, all animals were diligently monitored for mortality, body weight, body temperature, and clinical signs of distress. Immunization with three different doses of RV-1730 demonstrated complete protection against lethal infection (Figure 4A), with no observed clinical distress or loss of body weight in any of the immunized groups (Figure 4B; Supplementary Figures 5, 6). In contrast, the non-immunized control group exhibited severe clinical distress, leading to the euthanization of 90% of the mice due to high post-SARSCoV-2 challenge clinical severity scores (>10) (Figure 4B). Additional evaluation using a plaque reduction neutralization test (PRNT) against the SARS-CoV-2/human/ITA/INMI1/2020 (WT) virus showed significant titers of neutralizing serum antibodies at both 13 and 27 days post-prime immunization (Figure 4C). High-dose mRNA RV-1730 immunization (10 μg/mouse) induced a substantial level of neutralizing serum antibodies against SARS-CoV-2 after the prime immunization on day 0. Lower doses (1 μg/mouse and 5 μg/mouse) resulted in undetectable or low levels of neutralizing antibodies after the prime immunization but boosted immunizations (day 14) increased serum neutralizing antibody titers for all doses, with 5 μg/mouse and 10 μg/mouse doses yielding higher levels compared to the 1 μg/mouse dose. Prime-boost immunization at all three dose levels provided complete protection against lethal SARS-CoV-2 challenge and induced neutralizing serum antibodies. Histopathology analysis of lung tissue specimens indicated substantial reductions in total lung pathology and interstitial pneumonia, suggesting protection from the inflammatory effects of SARS-CoV-2 infection (Supplementary Figure 7). Overall, RV-1730 immunization in female K18-hACE2 mice provided robust protection against SARS-CoV-2 WT, inducing neutralizing antibodies and mitigating pathological effects, supporting its potential as an effective vaccine candidate.
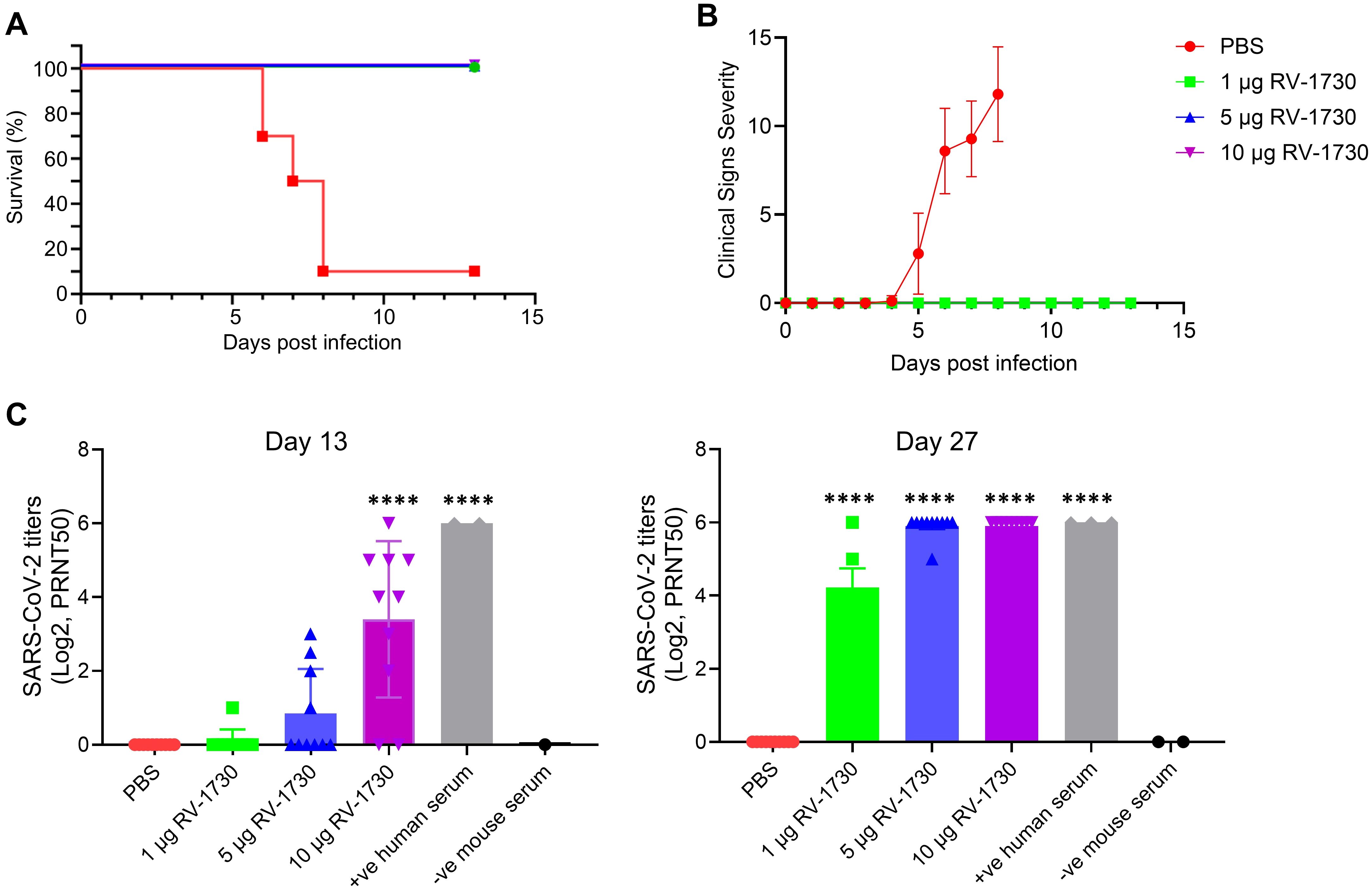
Figure 4. Protective efficacy of RV-1730 against SARS-CoV-2/human/ITA/INMI1/2020 in a K18-hACE2 mouse model of lethal infection. K18-ACE2 mice (n = 10 per group) were immunized with 3 doses of 1, 5, and 10 µg RV-1730 per mouse and challenged with wild type SARS-Cov-2 virus as described in “Methods” section. Mice in Group 1 were administered PBS, Group 2 mice were immunized with mRNA RV-1730 1 µg/animal, Group 3 mice were immunized with mRNA RV-1730 5 µg/animal, Group 4 mice were immunized with mRNA RV-1730 10 µg/animal before viral challenge. (A) Kaplan Meier curve of survival rate for mice challenged with SARS-CoV-2/human/ITA/INMI1/2020 virus. (B) clinical severity score curve in immunized and non-immunized mice after challenge with SARS-CoV-2 virus. (C) Plaque reduction neutralizing test of mouse serum against SARS-CoV-2/human/ITA/INMI1/2020. PRNT50 of post-prime immunization mouse serum collected on day 13 and day 27. Symbols and horizontal lines represent individual titers of each sample and mean titers of each group, respectively. Serum titers were expressed as reciprocals Log2 dilution. ****P<0.0001 compared to PBS (one-way ANOVA with Dunnett’s test).
Immunization of RV-1730 provides better protection against SARS-CoV-2 Delta challenge in mice than WT S2P vaccineSubsequently, we sought to compare the efficacy of S6P RV-1730 with that of the wild type S2P vaccine (WT S2P) equivalent to mRNA-1273. The protective potential of RV-1730 was assessed in female K18-hACE2 mice, with two different doses (0.5 μg/mouse and 5 μg/mouse) administered. Mice underwent prime immunization on day 0 and a booster shot on day 14, receiving either 0.5 μg/mouse or 5.0 μg/mouse of RV-1730 or WT S2P that has the same sequence as the mRNA-1273. On day 28, intranasal challenges were conducted with 1.5 x 104 PFU/mouse SARS-CoV-2/Delta variant. Prime-boost immunization with either 0.5 μg/mouse or 5.0 μg/mouse of RV-1730 and 5.0 μg/mouse of WT S2P vaccine conferred complete protection against SARSCoV-2 Delta (Figure 5A), preventing mortality, clinical distress, and weight loss (Figure 5A; Supplementary Figure 8). In contrast, the non-immunized control group exhibited distress, body weight loss in all mice, and necessitated euthanization due to the severity of post-SARS-CoV-2 Delta challenge clinical distress. Post-challenge viral loads were detected in the non-immunized group and the group immunized with 0.5 μg/mouse WT S2P but not in mice immunized with either dose of RV-1730 or the 5.0 μg/mouse dose of WT S2P in lung and nose (Figure 5B). Immunization with either dose of RV-1730 or 5.0 μg/mouse of WT S2P resulted in a dose-dependent increase in neutralizing titers against SARS-CoV-2/Delta in serum collected on days 13 and 27 (Figure 5C). In contrast, prime-immunization and boosting with PBS control or 0.5 μg/mouse of WT S2P vaccine resulted in undetectable or lower levels of neutralizing activity in both day 13 and day 27 sera (Figure 5D). The higher neutralizing activity in day 27 sera correlated with the 14-day survival outcome of the challenged mice (Figure 5A). Histopathology analysis of lung tissue specimens revealed an increased total lung pathological score, interstitial pneumonia, and perivascular cuffs in the PBS control group and the group immunized with 0.5 μg/mouse WT S2P vaccine compared to the groups immunized with RV-1730 vaccine or 5.0 μg/mouse WT S2P vaccine, both in the survival arm of the study and in the lungs of mice (n=4~5) euthanized on day 3 post-challenge (Supplementary Figure 9). All together, these findings indicate that S6P RV-1730 immunization provides superior protection against SARS-CoV-2 Delta challenge in mice compared to the WT S2P vaccine.
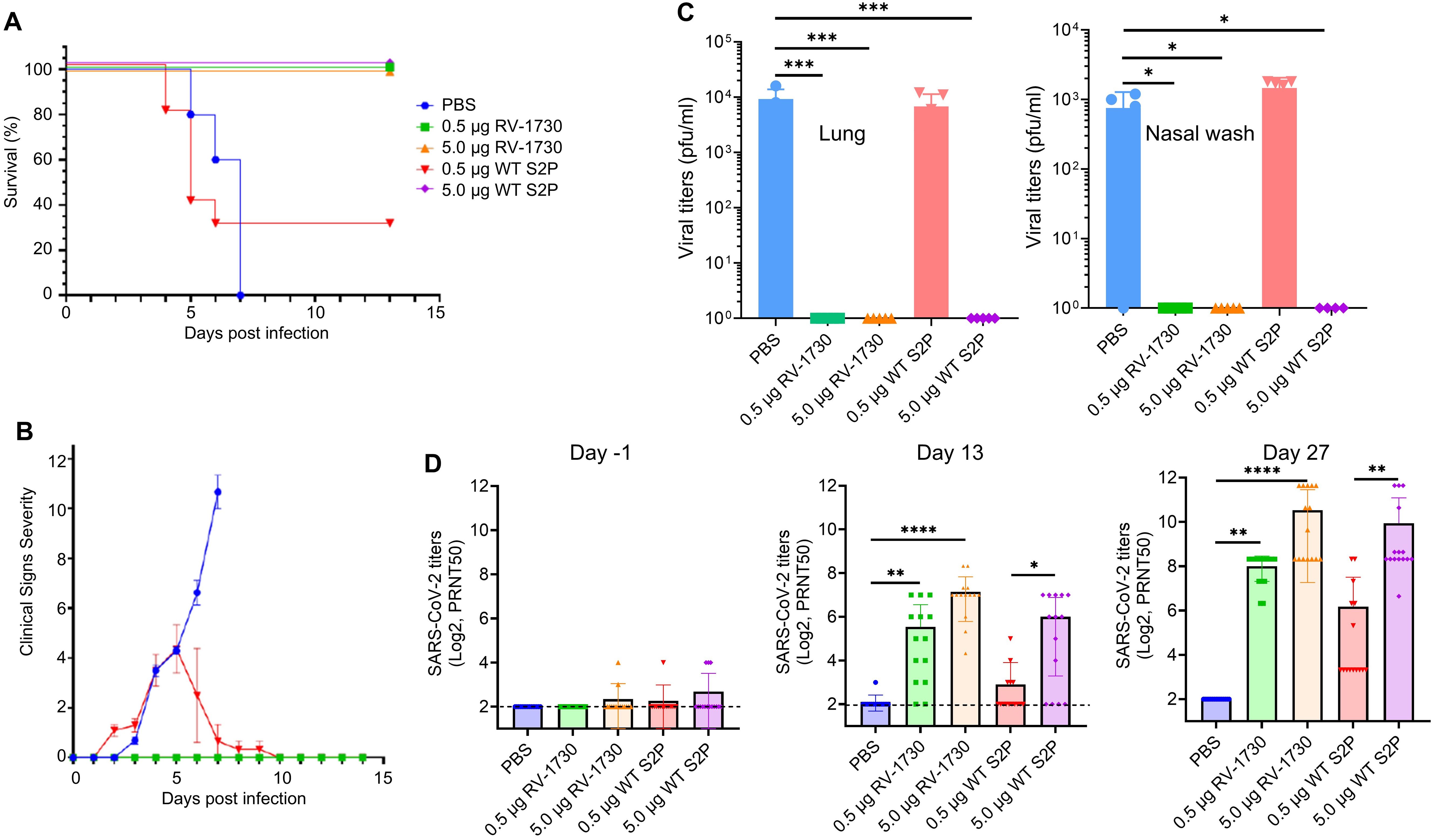
Figure 5. Protective efficacy of RV-1730 against SARS-CoV-2 B.1.1617.2 Delta variant/in a K18-hACE2 mouse model of lethal infection compared with WT S2P. (A) Survival rate for mice challenged with SARS-CoV-2 B.1.1617.2 Delta variant (n=10). Group 1 PBS control. Groups 2 and 3 – immunized with 0.5 µg or 5.0 µg RNAImmune mRNA vaccine, respectively. Groups 4 and 5 immunized with 0.5 µg or 5.0 µg WT S2P control mRNA, respectively. Kaplan Meier curve. (B) Clinical Severity Score Curve in immunized and non-immunized mice after challenge with SARS-CoV-2 B.1.1617.2 Delta Variant. (C) Post-SARS-CoV-2 viral load in lung tissue and nasal washes (n=5). Viral load in lung tissues and nasal washes harvested day 3 post-infection determined by plaque assay (left), viral load in nasal washes collected day 3 post-infection determined by plaque assay (right). Symbols and horizontal lines represent individual titers of each mouse and mean of each group, respectively. Any value less than 1 x 101 PFU/ml in (left) and any value less than 1 x 102 PFU/ml in (right) was considered as 0. (D) Mouse serum titration against SARS-CoV-2/Delta (n=15). PRNT50 of mouse serum collected on day -1 (left), day 13 (middle), and day 27 (right)post-boost immunization. *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001 (one-way ANOVA with Dunnett’s test).
Heterologous vaccination with RV-1730 provides benefits after primary vaccination with BNT162b2 and mRNA-1273Next, we aimed to assess the immunogenicity of sera resulting from both homologous and heterologous vaccinations and to evaluate functional neutralizing responses using a pseudovirus assay against various SARS-CoV-2 variants. Female Balb/c mice received intramuscular vaccinations with homologous (Group 1-3) and heterologous (Group 4 and 5) doses of BNT162b2, mRNA-1273, and RV-1730 at 5 µg per mouse, administered twice at 21-day intervals (Figure 6A; Supplementary Figure 10). Reciprocal serum endpoint IgG titers were determined through ELISA, employing a 2-fold serial dilution of the immunized sera. Prime-boost vaccination with homologous doses of BNT162b2, mRNA-1273, and RV-1730 generated robust IgG titers against the WT, Delta, BA.1, and BA,2 receptor-binding domain (RBD) of SARS-CoV-2 S (Figure 6B). Notably, homologous immunization with RV-1730 (group3) at day 35 post-immunization significantly increased IgG titers compared to either homologous immunization with BNT162b2 or mRNA-1273 against WT, Delta RBD (P < 0.001) (Figure 6A). In particular, heterologous immunization with RV-1730 in group 4 and 5 showed slightly an increased IgG titer against Delta as well as BA.2 RBD compared to those of homologus immunization of BNT162b2 and mRNA-1273. In pseudovirus neutralizing assays, serum samples derived from the Group 3, homologous immunization of RV-1730 effectively blocked infection from different variants (WT D614G, Delta, Omicron BA.1 and BA.2) to HEK293-ACE2 cells at very low concentrations compared to those of other groups derived from either homologus(Group1 and 2) or heterologous(Group 4 and 5). At 35 days post-immunization, the serum displayed strong neutralizing activity against all tested variants, with a range of 0.02-0.18% serum of IC50 (Figure 6C). These findings indicate that RV-1730 induces a strong and broadly neutralizing immune response, effectively inhibiting multiple SARS-CoV-2 variants—including WT D614G, Delta, and Omicron (BA.1 and BA.2)—even at low serum concentrations.
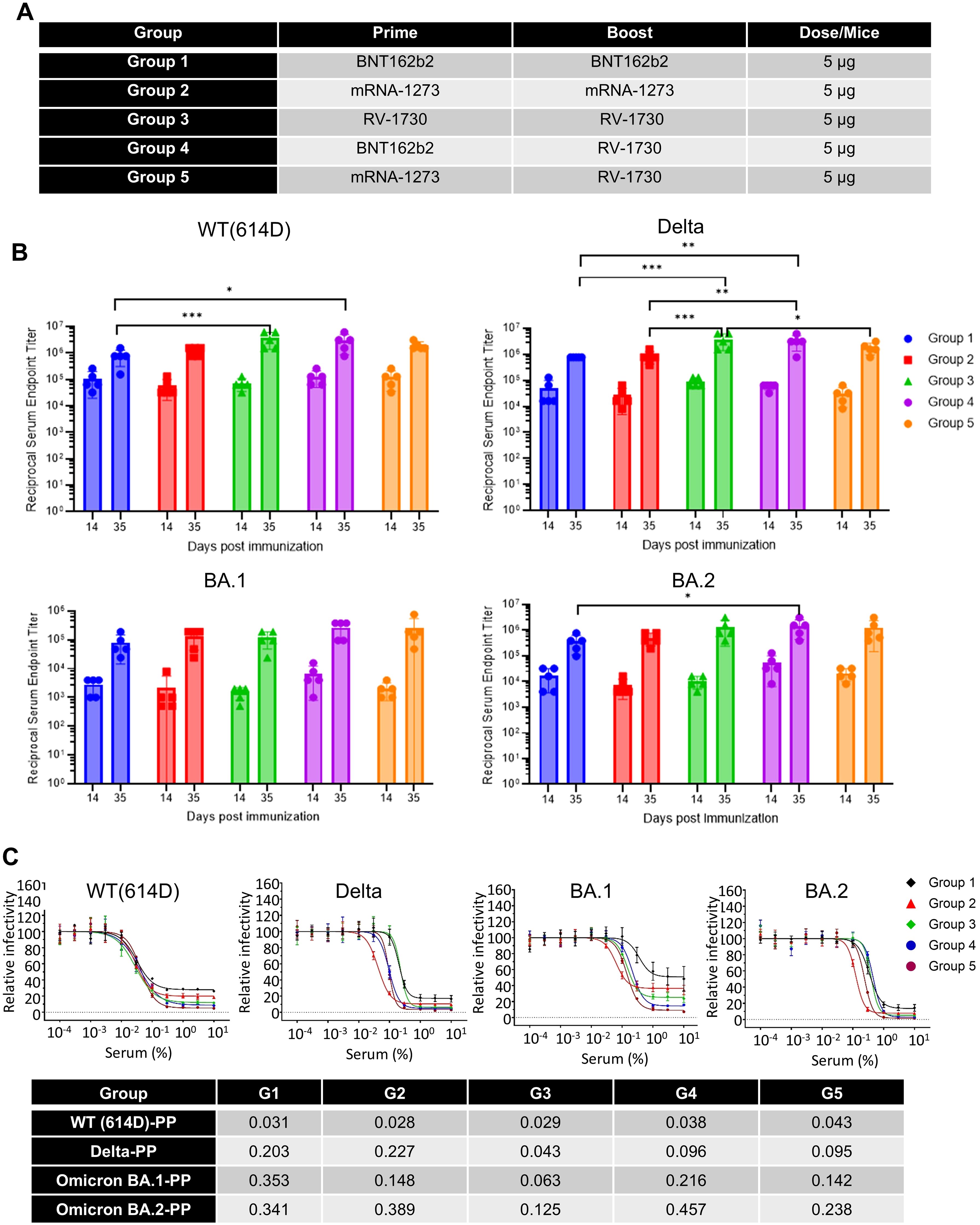
Figure 6. IgG and neutralization titers of homologous and heterologous SARS-CoV-2 spike mRNA-LNP immunized sera. (A) Homologous and heterologous immunization with different vaccines. (B) Endpoint IgG titers were determined for homologous and heterologous SARS-CoV-2 spike mRNA-LNP immunized sera against distinct variant spike RBD proteins. Mice (n=5) received prime immunization with SARS-CoV-2 spike mRNA-LNP (BNT162b2, mRNA-1273, and RV1730, 5 μg/mouse) and boost immunization either with the same vaccine (homologous) or a combination of vaccines (heterologous) three weeks after the prime immunization. Serum samples were collected on day 14 and 35 days to assess antibody levels specific to SARS-CoV-2 WT, Delta, Omicron BA.1 and BA.2 RBD proteins through ELISA. (C) The neutralization antibody titer against SARS-CoV-2 pseudovirus particles was analyzed, with the x-axis representing serum concentration and the y-axis indicating relative pseudoparticle infectivity. Different pseudovirus particles (PP), including 614D-PP, Delta-PP, Omicron BA.1-PP, and BA.2-PP, were used in the assay. The bottom table presents the IC50 (serum %) of heterologous vaccination sera in the pseudovirus neutralizing assay. *P<0.05, **P<0.01, and ***P<0.001 (two-way ANOVA with Tukey’s test).
The booster vaccination of commercial primary vaccines with RV-1730 elicits robust immune responsesHomologous and heterologous vaccinated mice received a booster shot with BNT162b2, mRNA-1273, and RV-1730 at day 72 post 2nd immunization. Blood samples were collected before each immunization at 14, 35, 49, 63, 77, and 105 days after the initial immunization (Figure 7A; Supplementary Figure 10). In booster immunization, both homologous and heterologous immunization induced strong IgG endpoint titers against SARS-CoV-2 S S1 of WT. Although heterologous booster immunization with BNT162b2 and RV-1730(Group 4) at day 105 significantly increased IgG titers against WT S1 compared to homologous immunization (Group 1, P < 0.05and Group 2, P <0.001) (Figure 7B). Only primary and booster vaccination with RV-1730 demonstrated a potent neutralizing antibody response against Omicron BA.2 and BA.5 (Figure 7C). RV-1730 exhibited approximately a 10-fold increase in neutralizing activity against Omicron BA.1 and BA.5 compared to sera before booster immunization. Additionally, homogenous immunization with RV-1730 as a booster showed a strong induction of IFN-γ+ CD8 T cell response by Delta S1 peptide pool compared to homogenous boosters with BNT162b2 and mRNA-1273 (Figures 8A, B). Furthermore, heterogenous boosting with RV-1730 following primary vaccination with BNT162b2 and mRNA-1273 enhanced the induction of IFN-γ+ CD8 T cell response by Delta S1 peptide pool (Figure 8C). Given that RV-1730 monovalent booster immunization exhibited comparable neutralizing activity to BA.5 primary vaccination, the variant-modified booster vaccination with RV-1730 can provide significantly higher neutralization titers against a diversity of current (e.g., BA.5) and historic SARS-CoV-2 variants compared to ancestral-based boosters. The enhanced responsiveness observed with RV-1730 may be influenced by the use of stimulating peptides that are homologous to the vaccine’s Delta variant sequence. In contrast, BNT162b2 and mRNA-1273, which are based on the ancestral strain, may show reduced responsiveness in this assay due to the heterologous nature of the peptides used (Figure 8C). In conclusion, booster immunization with RV-1730 not only elicited robust IgG responses against diverse SARS-CoV-2 variants but also demonstrated superior T cell activation, highlighting its potential as an effective and comprehensive vaccination strategy.

Figure 7. Serological evaluation of the booster vaccination of commercial primary vaccines with RV-1730. (A) Booster immunization using different vaccines in homologous and heterologous vaccinated mice. (B) Endpoint IgG titers against wild-type spike S1 proteins were measured on day 105 in sera from booster-immunized mice. Mice (n=5) were initially immunized with SARS-CoV-2 spike mRNA-LNP vaccines (BNT162b2, mRNA-1273, or RV1730) and received booster doses either with the same vaccine (homologous, Groups 1-3) or a combination of different vaccines (heterologous, Group 4) on day 0 (5 μg/mouse), day 21 (5 μg/mouse), and day 91 (2.5 μg/mouse), as detailed in Supplementary Figure 10. Serum samples collected on day 105 were analyzed for SARS-CoV-2 RBD-specific antibody levels using ELISA. *P<0.05, **P<0.01, and ***P<0.001 (two-way ANOVA with Tukey’s test). (C) The neutralizing antibody titer against SARS-CoV-2 pseudovirus particles is shown. The X-axis represents compound concentration, while the Y-axis indicates relative pseudovirus particle (PP) infectivity. The neutralizing activity of booster (third dose) vaccinated sera against BA.2 and BA.5-PP was assessed on day 105. The table below displays the IC50 values for heterologous vaccination sera in the pseudovirus neutralization assay.

Figure 8. T-cell responses in mice vaccinated with RV-1730 booster administration. (A) IFN-γ ELISPOT analysis was conducted on splenocytes. Two weeks after the administration of the third booster shot, splenocytes were harvested from five mice per group. These splenocytes (2 x 104 cells per 96-well plate) were then seeded onto mouse IFN-γ ELISpot plates (Mabtech) and ex vivo re-stimulated with pools of overlapping peptides derived from the SARS-CoV-2 Delta Spike for a duration of 16 hours. Subsequently, images were captured and quantified using Cytation7. Presented is a representative image of the ELISpot. (B) The quantitative analysis of the ELISpot data revealed statistical significance. (C) CD4/CD8 T cell analysis. The splenocytes were isolated and activated following the procedures outlined in the ELISPOT assays. The upper row illustrates the gating steps for CD3+ CD4+ and CD3+ CD8+ cell populations. IFN-γ+ and IL-4+ within the CD4 and CD8 subpopulations were analyzed by a flow cytometer. The percentage of CD4/IFN-γ, CD4/IL-4, CD8/IFN-γ, and CD8/IL-4 cells are presented. *P<0.05, **P<0.01, and ****P<0.0001 (one-way ANOVA
留言 (0)