
記住我
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by multi-organ and multi-system involvement (1). SLE complicated with thrombotic microangiopathy (TMA) and non-cirrhotic portal hypertension (NCPH) is rare in clinical practice. TMA is a kind of clinicopathologic syndrome with microangiopathic hemolytic anemia, thrombocytopenia, and organ damage mainly based on terminal arteriole and capillary endothelial injury and thrombosis (2). The pathological manifestations include extensive microvascular endothelial cell swelling, microthrombus formation, and capillary cavity stenosis. NCPH is a rare vascular liver disease with clinical manifestations of portal hypertension without cirrhosis or severe fibrosis. It is reported that NCPH is related to autoimmune diseases; however, only a few cases have been reported in SLE. Here, we report a case of a female patient with SLE complicated by TMA and NCPH.
2 Case descriptionA 53-year-old woman presented with confusion for 6 h. Physical examination revealed a blood pressure of 183/110 mmHg, oxygen saturation of 96%, body temperature of 36.2°C, heart rate of 92 beats/min, and respiratory rate of 16 breaths/min. She also presented with drowsiness, a pale face, and speech confusion. Her superficial lymph nodes, liver, and spleen were not enlarged. Her muscle tone was weak, and muscle strength was diminished. The meningeal stimulation sign was negative. The patient had no notable past medical history. Two months prior, she had experienced dry mouth and occasional gum bleeding, for which she was evaluated at a local hospital. Routine blood work, biochemical tests, and cancer markers were normal, and bone marrow cytology showed no abnormalities. She also had no significant family history or history of heavy alcohol consumption or cigarette smoking.
The laboratory data on admission are as follows. Routine laboratory examinations showed the following: hemoglobin level, 6.4 g/dL [normal range (NR), 11.5–15 g/dL]; total leukocyte count, 2.52×109/L (NR, 4–10×109/L); and platelet count, 26×109/L (NR, 125–350×109/L). Her C-reactive protein level was <6 mg/L (NR, 0–10 mg/L), and erythrocyte sedimentation rate was 156 mm/h (NR, 0–15 mm/h). Further laboratory testing showed high levels of brain natriuretic peptide (BNP; 3,830.70 pg/mL; NR, 0–100 pg/mL), troponin I (0.052 ng/mL), serum lactate dehydrogenase (LDH; 475.09 IU/L; NR, 100–240 IU/L), creatinine (132.70 µmol/L; NR, 45–84 µmol/L), and creatine kinase isoenzyme (56.00 U/L; NR, 0–24 U/L). Her 24-h urinary protein level was 782.99 mg. She tested positive for a direct anti-human globulin test but negative for an indirect anti-human globulin test. A peripheral blood smear showed 0.5% schistocytes. The patient was strongly positive for anti-nuclear antibody, anti-SSA, anti-SSB, anti-RO52, rheumatoid factors, and anti-double-stranded DNA but negative for anti-Smith, anti-cardiolipin, anti-β2 glycoprotein type I, anti-cyclic citrullinated peptide, HLA-B27, anti-myeloperoxidase, anti-protease 3, anti-glomerular basement membrane antibodies, and lupus anticoagulant. Complement C3 and C4 levels were low [0.29 g/L (NR, 0.90–1.80 g/L) and 0.05 g/L (NR, 0.10–0.40 g/L), respectively]. Electrocardiography indicated ST segment changes in some leads, partial lead T wave changes, and a prolonged Q–T interval. Abdominal computed tomography (CT) showed splenomegaly, whereas the size and structure of both kidneys were within normal limits.
SLE was diagnosed according to the American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) classification criteria of 2019 for SLE (3). The SLE disease activity index 2000 (SLEDAI-2K) was 18, indicating severe SLE activity. She was initially treated with a high dose of methylprednisolone (500 mg) for 3 days and gamma globulin (0.4 g/kg/day) for 5 days, followed by immunotherapy with methylprednisolone (60 mg/day). Immunosuppressants such as mycophenolate, cyclophosphamide, calcineurin inhibitors (e.g., cyclosporine, tacrolimus), and azathioprine were viable treatment options for systemic lupus erythematosus involving the hematologic system. Because the patient had significantly reduced total leukocyte count, cyclophosphamide was prone to cause bone marrow suppression, and based on our previous treatment experience, we selected the calcineurin inhibitor tacrolimus(1 mg twice daily). By the 7th day, the patient’s neurological symptoms had completely resolved. Laboratory results showed a total leukocyte count of 6.88×109/L, a hemoglobin level of 9.4 g/dL, a platelet count of 56×109/L, a BNP level of 1691.60 pg/mL, and a troponin I level of 0.027 ng/mL.
On the 30th day, the patient gradually developed dizziness, weakness, and muscle soreness, accompanied by lower limb edema and shortness of breath after activity. CT showed high-density imaging in the upper lobe of the right lung (new), possibly indicating inflammation, bilateral pleural effusion (new), or abdominal effusion. The patient tested negative for the 2019nCoV RdRP gene. The patient was administered ceftriaxone combined with SMZ for infection prevention, methylprednisolone (60 mg) for anti-inflammation, tacrolimus (1 mg, twice daily) for immune suppression, diuretic for cardiac load reduction and blood pressure control, and low-molecular-weight heparin (2,500 U every 12 h) for anticoagulation. Neurological symptoms partially resolved. Re-examination of laboratory indicators showed the following: hemoglobin level, 94 g/L; platelet count, 86×109/L; BNP level, 230 pg/mL; and troponin I level, 0.025 ng/mL. The tacrolimus drug concentration was 1.9 ng/mL. Chest CT showed a decrease in pulmonary infection. On the 35th day, because of abnormal liver function, tacrolimus was replaced with mycophenolate (0.75 g twice daily).
On the 42nd day, the patient experienced dizziness and aggravated muscle pain accompanied by low consciousness, slow response, and an inability to understand and complete simple movements. BNP, troponin I, creatinine, and myocardial enzyme levels were all higher than before. The complement level was lower, and the erythrocyte sedimentation rate was higher. She had a SLEDAI score of 18, which indicated severe lupus activity. She was again treated with high-dose methylprednisolone (500 mg) for 3 days and immunoglobulin (0.4 g/kg/day) for 5 days. During the treatment, the patient’s mental symptoms improved, and she was able to understand and complete motor commands. On the 47th day, the patient experienced a recurrence of confusion and delayed responses. Re-examination showed decreased hemoglobin and platelet levels compared with previous values, with peripheral schistocytes at approximately 6.5%. ADAMTS13 activity was 49.18%, ADAMTS13 inhibitor was negative, LDH and creatinine levels increased, and troponin I level increased to 0.087 ng/mL. Abdominal color ultrasonography revealed a large amount of ascites, diffuse liver lesions, splenomegaly, and varicose splenic dilation (Figure 1). Enhanced CT revealed diffuse liver disease along the portal veins, intrahepatic lymphatic dilatation, esophageal and gastric varices, a splenorenal vein shunt, and splenomegaly (Figure 1). The patient underwent peritoneal puncture and catheterization to drain the turbid yellow ascites. The bacteriological results for the ascites were negative, and cytological pathological examination of the ascites showed no malignant changes. The patient was negative for the spectrum of autoimmune liver disease antibodies, hepatitis virus, tumor markers, and ceruloplasmin. SLE complicated with TMA, and NCPH was considered. She was administered rituximab (500 mg) for immunosuppressive therapy, and mycophenolate was discontinued. Considering the possibility of complement-mediated TMA (CM-HUS) or other atypical HUS, plasma exchange was considered, as it can remove abnormal complement regulatory proteins, CFH antibodies, and other pathogenic factors while simultaneously supplementing normal complement regulatory proteins. The patient underwent plasma exchange (seven times, with 2,000 mL of plasma each time). Low-molecular-weight heparin dosage was increased to 5,000 U every 12 h for anticoagulation. On the 48th day, the 2019nCoV RdRP gene was positive, and nematavir/ritonavir was administered for novel coronavirus treatment. Lung infection lesions increased (Figure 1). She was treated with piperacillin sodium/tazobactam sodium for infection, albumin supplementation, diuresis, methylprednisolone (60 mg) for inflammation, and internal environment stability. On the 59th day, the patient’s symptoms and laboratory indices improved (Figure 2). Laboratory examinations showed a total leukocyte count of 4.32×109/L, a hemoglobin level of 9.6 g/dL, a platelet count of 155×109/L, a BNP level of 62.40 pg/mL, and a troponin I level of 0.012 ng/mL. C-Reactive protein was <6 mg/L, erythrocyte sedimentation rate decreased to13 mm/h, serum lactate dehydrogenase was 260.46 IU/L, creatinine was 106.00 umol/L, and creatine kinase isoenzyme was 28.00 U/L. The proportion of peripheral schistocytes decreased to 0.2%. She was discharged to the local hospital for continued treatment. At discharge, her medications included prednisone 50 mg/day, aspirin 75 mg/day, pantoprazole 40 mg/day, and calcium carbonate 0.6 g/day, all taken orally. Rituximab (500 mg) was administered again 2 weeks post-discharge after her COVID-19 test turned negative. Six months later, during follow-up, her prednisone dose had been gradually reduced to 10 mg/day, with no recurrence of mental symptoms, normal hemoglobin and platelet levels, and complete ascites resolution.
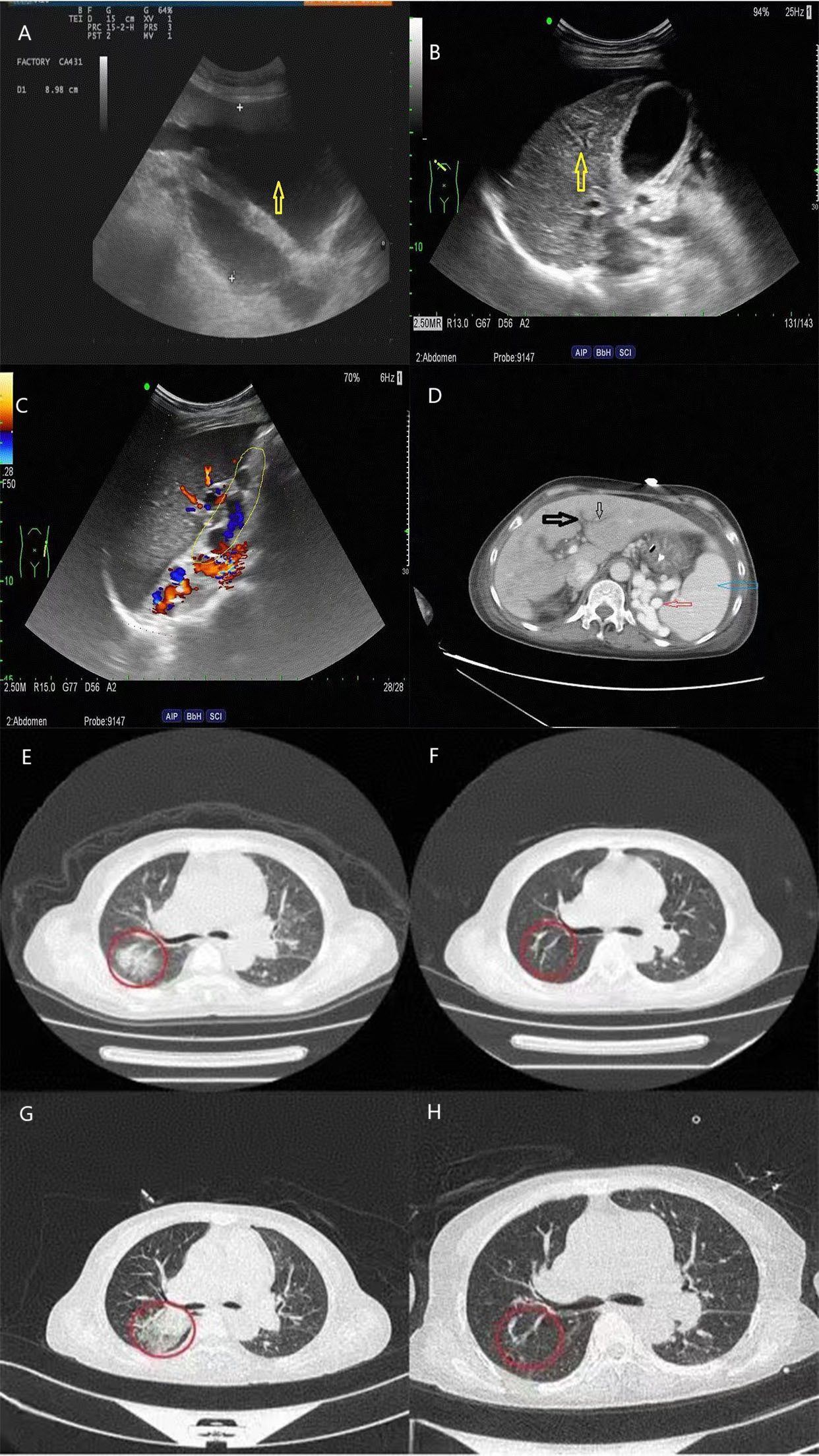
Figure 1. Abdominal ultrasonography and abdominal/chest CT scans of the patient. (A) Abdominal color ultrasonography indicates excessive ascites; (B) diffuse parenchymal thickening of the liver; (C) splenic veins are tortuous and dilated; (D) the black arrows show intrahepatic lymphatic dilatation; the blue arrow shows splenomegaly; and the red arrow shows collateral circulation; (E–H) show changes in chest CT infection lesions over time.
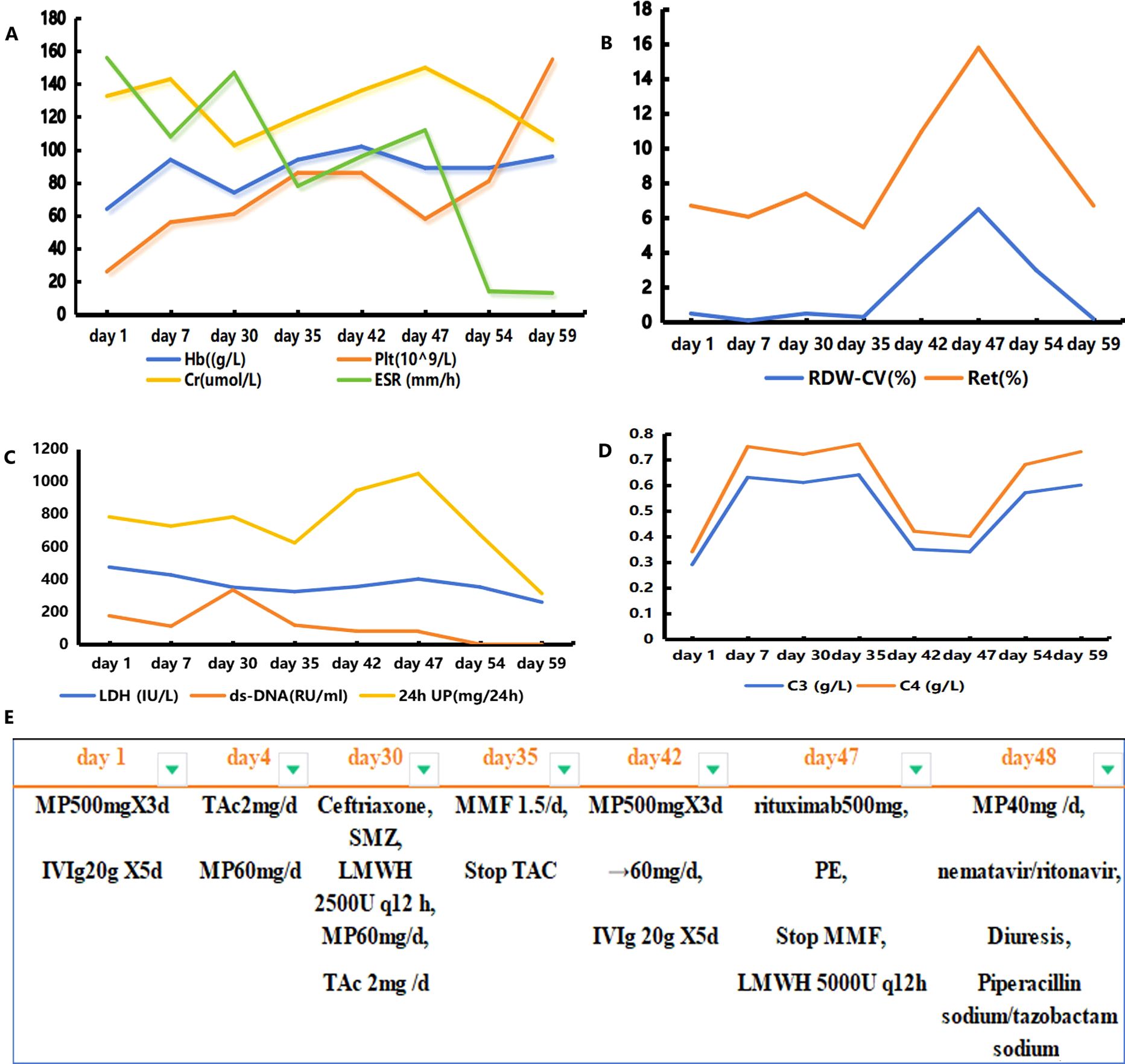
Figure 2. Graph illustrating the evolution of key laboratory parameters and the treatments administered. Panels (A–D) represent the changes in the most relevant laboratory parameters over time; panel (E) depicts the different treatments administered to the patient. Hb, hemoglobin; Plt, platelet count; RDW-CV, red cell distribution width coefficient of variation; Ret, reticulocyte ratio; Cr, creatinine; ESR, erythrocyte sedimentation rate; 24-h UP, 24-h urinary protein; MP, methylprednisolone; IVIg, gamma globulin; TAc, tacrolimus; MMF, mycophenolate; PE, plasma exchange; LMWH, low-molecular-weight heparin.
3 DiscussionWe identified some clinical issues from the case report. First, infection may be a precursor to SLE disease activity leading to TMA. Chen et al. (4) reviewed the clinical data of 25 patients with SLE complicated with TMA. Among them, 22 patients had moderate-to-severe SLE activity at the time of TMA, suggesting that TMA was mostly parallel to the disease activity of their primary condition. Additionally, 16 patients had concurrent infections, suggesting that infection may be an inducing factor. In patients with SLE, infection can cause TMA either directly or indirectly by inducing lupus activity, with TTP, APS, and complement-mediated TMA being the most relevant causes. However, TMA in lupus patients can also be caused by infection, medications (particularly because of calcineurin inhibitor toxicity), malignant hypertension, or malignancy (5, 6). Calcineurin inhibitors (such as cyclosporine and tacrolimus) can induce dose-dependent endothelial dysfunction, potentially leading to TMA. Although tacrolimus can cause TMA, the patient in this case showed symptoms and laboratory improvement following initial treatment with tacrolimus. On day 35, tacrolimus was discontinued because of abnormal liver function, and mycophenolate was introduced as a replacement. The patient’s condition worsened after discontinuing tacrolimus, but given the low tacrolimus concentration, it is unlikely that tacrolimus induced TMA in this case. In addition, malignant hypertension can cause renal endothelial cell damage, leading to TMA (6). Although the patient had elevated blood pressure at admission, it was stable at the time of clinical worsening, suggesting that malignant hypertension was unlikely to be the primary cause of TMA. Infections are considered the primary cause of lupus activity. In addition to the common Shiga toxin-producing Escherichia coli-associated hemolytic uremic syndrome, TMA associated with infections such as those of human immunodeficiency virus, cytomegalovirus, and novel coronavirus has been reported (7). This may be related to direct damage to endothelial cells, complement-mediated damage, or other factors (7). In our case, during the onset of the disease, treatment was effective, but the patient’s symptoms and test indicators worsened after COVID-19 and bacterial lung infections. Her condition did not improve after treatment with high-dose steroids combined with gamma globulin. TMA was considered because of the combination of decreasing hemoglobin and platelet levels, increased LDH and creatinine levels, and a peripheral schistocyte proportion of approximately 6.5%. Infection might have been the inducing factor for the progression of her disease activity to TMA.
Second, patients with TMA and normal ADAMTS13 activity have a poor treatment response. The mechanisms of SLE complicated by TMA are complex and diverse and easily confused with lupus activity, which can impact diagnosis and treatment decisions. Kidney biopsy is the pathological gold standard for TMA diagnosis (8). However, when a kidney biopsy cannot be performed, TMA can be indicated by specific laboratory indicators, such as a decrease in hemoglobin level and platelet count and an increase in LDH level. Before a clinical diagnosis of SLE complicated by TMA, our patient was administered high-dose methylprednisolone combined with immunoglobulin therapy based on her severe lupus activity. However, she showed no improvement in thrombocytopenia, anemia, repeated mental abnormalities, or massive abdominal ascites. Abdominal color ultrasonography revealed disseminated liver lesions, spleen enlargement, and esophageal varicosities. The patient refused to undergo liver or kidney biopsy; however, her microangiopathic hemolytic anemia, thrombocytopenia, elevated LDH level, broken red blood cells on a peripheral blood smear, and portal hypertension indicated SLE combined with TMA. Microthrombus formation can block the hepatic vein or inferior vena cava in the upper segment of the liver, resulting in massive ascites and presenting as Budd–Chiari syndrome (BCS). Anticoagulation is the primary treatment for all cases of BCS in a significantly hypercoagulable state (9). The patient was treated with rituximab (500 mg) and plasma exchange seven times, with 2,000 mL of plasma exchanged each time. The patient’s neuropsychiatric symptoms and laboratory indicators improved. ADAMTS13 activity was normal. ADAMTS13 inhibitor and antiphospholipid antibodies were negative. What type of antibodies could be involved? We suggest that the antibodies may be anti-endothelial antibodies; however, relevant testing was not conducted in this case. We hope that future studies will address similar issues in greater depth and that more scholars will investigate this topic further. The antibodies may be associated with vascular endothelial cell damage induced by SLE, in vivo inflammation, and the activation of TMA through complement or other immune pathways. Additionally, SLE-induced dysfunction of the coagulation system may contribute to the occurrence and progression of TMA despite normal ADAMTS13 activity (10). Furthermore, infection could prompt a pro-inflammatory state, activating the clotting and complement cascades, potentially inducing TMA. Patients with TMA secondary to SLE are heterogeneous, and normal ADAMTS13 activity indicates a poor prognosis (11). Plasma exchange can remove cytokines, toxins, and autoantibodies; replace defective plasma factors, and improve TMA symptoms and prognosis in SLE patients. However, the case fatality rate remains high at 27.85%–62.5% (12). Rituximab is an antibody against CD20 that binds to the surface of B cells and eliminates B cells through complement and antibody-dependent cellular cytotoxicity, thereby inhibiting the excessive release of cytokines, reducing endothelial cell damage, and reducing the production of vWF (13). Current guidelines suggest rituximab as a second-line treatment for TMA (14). Furthermore, rituximab can reduce treatment duration and prevent subsequent recurrence (15). From a literature review (2, 11, 16–21), 24 patients with SLE complicated by TMA were treated with rituximab, and most had acute kidney injury as the first symptom; however, in this case, confusion was the initial symptom. Most patients involve only the kidney and nervous system. In this case, the nervous system, blood, kidney, and heart were simultaneously involved, indicating a serious condition and poor prognosis. Rituximab therapy has shown good results, mostly with two or more standard doses combined with high-dose steroids and plasma exchange (Table 1). The patient’s peripheral blood B cells were cleared after treatment with 500 mg rituximab, and she demonstrated a positive response to rituximab combined with plasma exchange treatment.
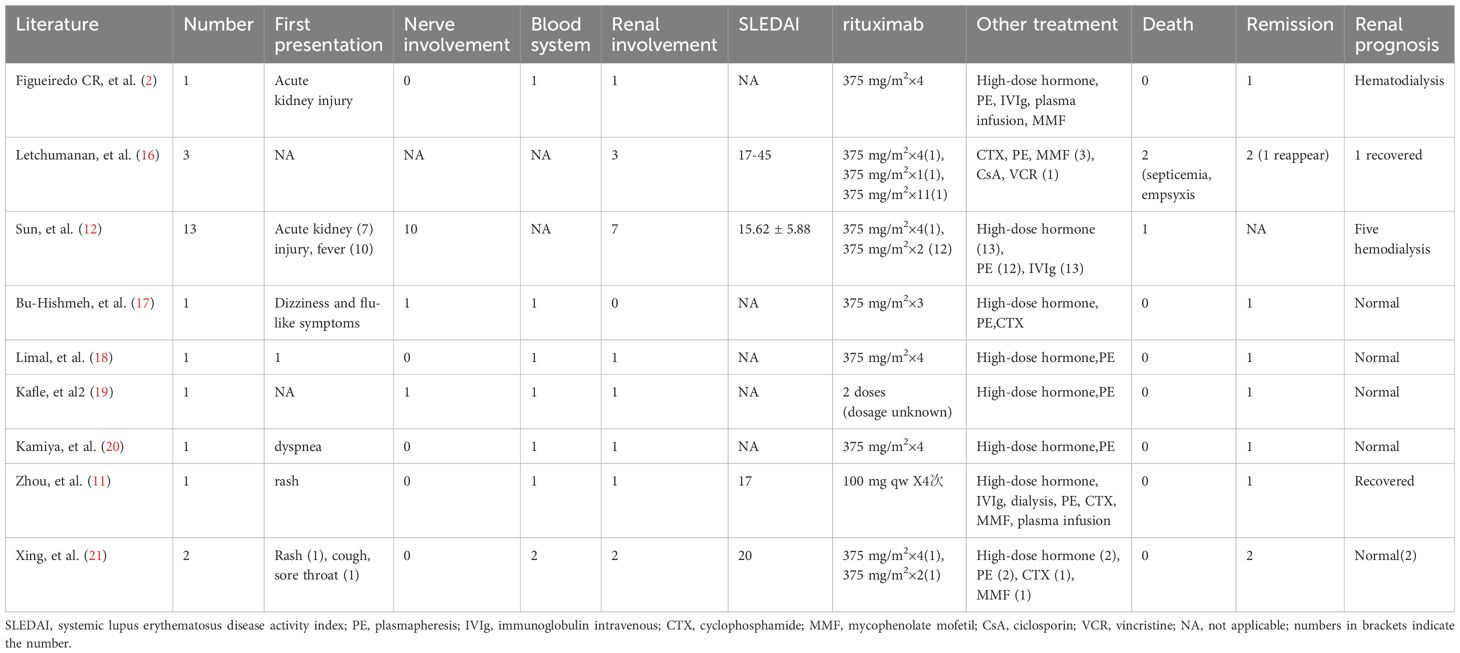
Table 1. Summary of patients with TMA secondary to SLE treated with rituximab.
Third, portal hypertension can deteriorate concurrently with SLE exacerbation. Table 2 summarizes the immunosuppressive treatment of NCPH secondary to SLE over the past 25 years (22–28). The age of onset ranged from 19 to 48 years, with most patients being women (six out of seven). Although it took a considerable time for NCPH to manifest as ascites, its progression was rapid once it did. In this instance, NCPH manifested a correlation with SLE activity. The cause of NCPH in SLE remains unidentified; however, several pathophysiological mechanisms have been hypothesized, including immune alterations and hypercoagulability (22). This case suggests that autoimmunity and thrombosis could underlie the development of SLE-associated NCPH. We think that the NCPH and the cerebral and myocardial alterations can be attributed to microangiopathic damage. Hence, some authors suggest steroids as a possible treatment option. In our case, the simultaneous deterioration of SLE hindered glucocorticoid dose reduction. Supplementing anticoagulants with enhanced immunosuppressive therapy may improve the management of portal vein thrombosis, and amelioration of splenomegaly and esophageal varices has been reported after immunosuppressive therapy for SLE. However, every instance occurred in individuals who experienced a brief interval between the onset of SLE and the diagnosis of NCPH (Table 2). In our case, at disease onset, the abdominal CT scan revealed an enlarged spleen with no evidence of ascites or any abnormal liver structure. As SLE worsens, NCPH progresses rapidly. It is important to recognize the onset of NCPH early and provide prompt treatment. NCPH treatment includes diuretics, anticoagulants, oral calcium channel blockers, prostacyclin, endoscopic injection sclerotherapy, partial splenic embolization, splenectomy, and devascularization of the stomach vessels (Table 2). Most of these treatments improve when SLE is managed concurrently.
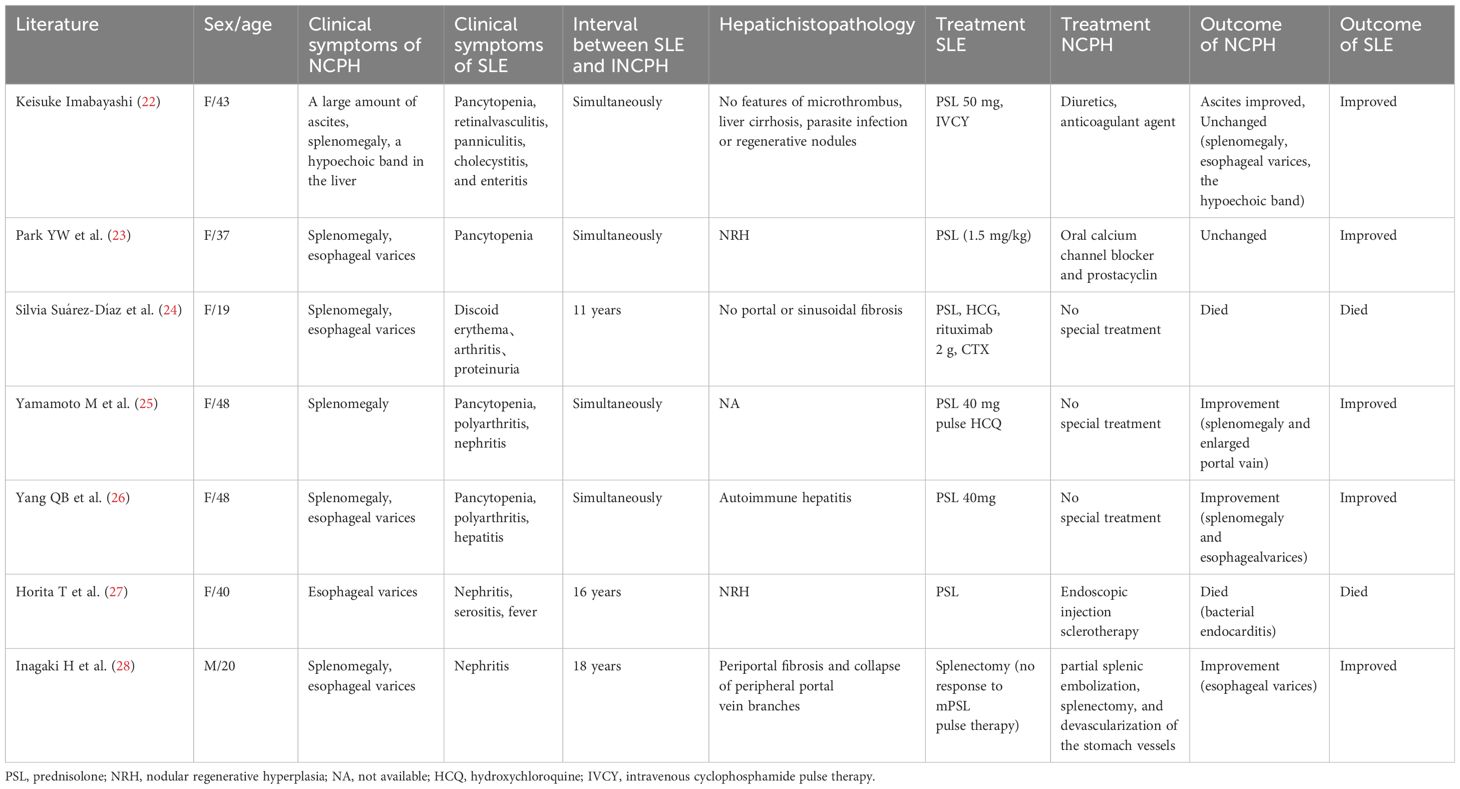
Table 2. Immunosuppressive treatment of SLE with NCPH reported in the past 25 years.
This case suggests that rituximab, combined with plasma exchange, anticoagulation, and diuretics, may be an effective therapeutic option for patients with SLE complicated by TMA and NCPH. However, the pathogenesis and prognosis of SLE in the context of TMA and NCPH remain complex.
Data availability statementThe original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Ethics statementThe studies involving humans were approved by Medical Ethics Committee of the Second Affiliated Hospital of Xiamen Medical College. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributionsJH: Writing – original draft, Writing – review & editing. WF: Supervision, Writing – review & editing. XC: Supervision, Writing – review & editing. SW: Investigation, Writing – review & editing. ZD: Data curation, Writing – review & editing. YZ: Investigation, Writing – review & editing. YL: Investigation, Writing – review & editing. PX: Data curation, Writing – review & editing.
FundingThe author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Xiamen Medical and Health Guidance Project (No. 3502Z20224ZD1251) and Xiamen Medical College university-level project (No. K2023-41).
AcknowledgmentsWe want to acknowledge the hard work and dedication of all the staff involved. We also appreciate the selfless dedication of the patient.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AbbreviationsSLE, systemic lupus erythematosus; TMA, thrombotic microangiopathy; NCPH, non-cirrhotic portal hypertension; HE, hematoxylin and eosin; MOC31, an antibody used in pathology; EMA, epithelial membrane antigen; CRM, calretinin marker; CD163, a marker for macrophages; PAS, periodic acid–Schiff; NR, normal range; BNP, brain natriuretic peptide; LDH, lactate dehydrogenase; CT, computed tomography; ACR, American College of Rheumatology; EULAR, European League Against Rheumatism; SLEDAI-2K, SLE Disease Activity Index 2000; Q–T interval, the time between the start of the Q wave and the end of the T wave in the heart’s electrical cycle; ADAMTS13, a disintegrin and metalloproteinase with thrombospondin motifs 13; COVID-19, coronavirus disease 2019; RdRP, RNA-dependent RNA, polymerase; BCS, Budd–Chiari syndrome; vWF, von Willebrand factor; APS, antiphospholipid syndrome; TTP, thrombotic thrombocytopenic purpura; HLA, human leukocyte antigen; SSA, Sjögren’s syndrome-related antigen A; SSB, Sjögren’s syndrome-related antigen B; IgG, immunoglobulin G; IgA, immunoglobulin A; IgM, immunoglobulin M; SMZ, sulfamethoxazole.
References1. Bensalek F, Joulal H, Yousfi J, Zahlane M, Benjilali L, Essaadouni L. Autoimmune myelofibrosis revealing a systemic lupus erythematosus. Eur J Case Rep Intern Med. (2024) 11:4511. doi: 10.12890/2024004511
PubMed Abstract | Crossref Full Text | Google Scholar
2. Figueiredo CR, Escoli R, Santos P, Sofia F, Lopes K. Thrombotic microangiopathy in a patient with systemic lupus erythematosus and anti-factor H autoantibodies. CEN Case Rep. (2022) 11:26–30. doi: 10.1007/s13730-021
PubMed Abstract | Crossref Full Text | Google Scholar
3. Del Río Zanatta H, Zambrano Zambrano A, Belmont Nava P, Lizbeth Puntos Guízar C, Martinez Salazar J. Comprehensive evaluation of neuropsychiatric and mucocutaneous manifestations in the diagnosis of systemic lupus erythematosus: a complete clinical approach to a case. Cureus. (2023) 15:e47380. doi: 10.7759/cureus.47380
PubMed Abstract | Crossref Full Text | Google Scholar
4. Ming-Han C, Ming-Huang C, Wei-Sheng C, Peter MC, Hui-Ting L, Hsiao-Yi L, et al. Thrombotic microangiopathy in systemic lupus erythematosus: a cohort study in North Taiwan. Rheumatology. (2011) 50:768–75. doi: 10.1093/rheumatology/keq311
PubMed Abstract | Crossref Full Text | Google Scholar
6. Kotzen ES, Roy S, Jain K. Antiphospholipid syndrome nephropathy and other thrombotic microangiopathies among patients with systemic lupus erythematosus. Adv Chronic Kidney Dis. (2019) 26:376–86. doi: 10.1053/j.ackd.2019.08.012
PubMed Abstract | Crossref Full Text | Google Scholar
7. De Fabritiis M, Angelini ML, Fabbrizio B, Cenacchi G, Americo C, Cristino S, et al. Renal thrombotic microangiopathy in concurrent COVID-19 vaccination and infection. Pathogens. (2021) 10:1045. doi: 10.3390/pathogens10081045
PubMed Abstract | Crossref Full Text | Google Scholar
8. Chen RY, Li XZ, Lin Q, Tang HY, Cui NX, Jiang L, et al. Pathological evaluation of renal complications in children following allogeneic hematopoietic stem cell transplantation: a retrospective cohort study. BMC Pediatr. (2023) 23:186. doi: 10.1186/s12887-023-03996-1
PubMed Abstract | Crossref Full Text | Google Scholar
9. Torun ES, Erciyestepe M, Yalçınkaya Y, Gül A, İnanç M, Öcal L, et al. A case of Budd-Chiari syndrome associated with antiphospholipid syndrome treated successfully by transjugular intrahepatic portosystemic shunt. Clin Med Insights Case Rep. (2022) 15:11795476221100595. doi: 10.1177/11795476221100595
PubMed Abstract | Crossref Full Text | Google Scholar
10. Lansigan F, Isufi I, Tagoe CE. Microangiopathic haemolytic anaemia resembling thrombotic thrombocytopenic purpura in systemic lupus erythematosus: the role of ADAMTS13. Rheumatol (Oxford). (2011) 50:824–9. doi: 10.1093/rheumatology/keq395
PubMed Abstract | Crossref Full Text | Google Scholar
11. Zhou YZ, Zhao JL, Cao XY, Zheng K, Wu QJ, Zeng XF. The 476th case: skin rash, edema, thrombocytopenia and anemia. Zhonghua Nei Ke Za Zhi. (2020) 59:250–2. doi: 10.3760/cma.j.issn.0578-1426.2020.03.019
PubMed Abstract | Crossref Full Text | Google Scholar
12. Sun F, Wang X, Wu W, Wang K, Chen Z, Li T, et al. TMA secondary to SLE: rituximab improvers overall but not renal survival. Clin Rheumatol. (2018) 37:213–8. doi: 10.1007/s10067-017-3793-4
PubMed Abstract | Crossref Full Text | Google Scholar
14. Fanouriakis A, Kostopoulou M, Cheema K, Anders HJ, Aringer M, Bajema I, et al. 2019 Update of the joint European League Against Rheumatism and European Renal Association-European Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations for the management of lupus nephritis. Ann Rheum Dis. (2020) 79:713–23. doi: 10.1136/annrheumdis-2020-216924
PubMed Abstract | Crossref Full Text | Google Scholar
16. Letchumanan P, Ng HJ, Lee LH, Thumboo J. A comparison of thrombotic thrombocytopenic purpura in an inception cohort of patients with and without systemic lupus erythematosus. Rheumatol (Oxford). (2009) 48:399–403. doi: 10.1093/rheumatology/ken510
PubMed Abstract | Crossref Full Text | Google Scholar
17. Abu-Hishmeh M, Sattar A, Zarlasht F, Ramadan M, Abdel-Rahman A, Hinson S, et al. Systemic lupus erythematosus presenting as refractory thrombotic thrombocytopenic purpura: a diagnostic and management challenge. A case report and concise review of the literature. Am J Case Rep. (2016) 17:782–7. doi: 10.12659/ajcr.898955
PubMed Abstract | Crossref Full Text | Google Scholar
18. Limal N, Cacoub P, Sène D, Guichard I, Piette JC. Rituximab for the treatment of thrombotic thrombocytopenic purpura in systemic lupus erythematosus. Lupus. (2008) 17:69–71. doi: 10.1177/0961203307083479
PubMed Abstract | Crossref Full Text | Google Scholar
19. Kafle P, Malakoff GL. Coexistence of systemic lupus erythematosus and thrombotic thrombocytopenic purpura: a case report. Tenn Med. (2012) 105:37–8.
PubMed Abstract | Google Scholar
20. Kamiya K, Kurasawa K, Arai S, Maezawa R, Hanaoka R, Kumano K, et al. Rituximab was effective on refractory thrombotic thrombocytopenic purpura but induced a flare of hemophagocytic syndrome in a patient with systemic lupus erythematosus. Mod Rheumatol. (2010) 20:81–5. doi: 10.1007/s10165-009-0231-8
PubMed Abstract | Crossref Full Text | Google Scholar
21. Xing BH, Yang Y, Li WB. Rituximab in the treatment of systemic lupus erythematosus complicated with thrombotic microangiopathosis in 2 cases. Chin J Rheumatol. (2023) 27:246–9. doi: 10.3760/cma.j.cn141217-20220215-00055
Crossref Full Text | Google Scholar
22. Imabayashi K, Nakano K, Iwata S, Tanaka Y. A case of systemic lupus erythematosus with marked ascites due to idiopathic non-cirrhotic portal hypertension. Mod Rheumatol Case Rep. (2021) 5:285–91. doi: 10.1080/24725625.2021.1904607
PubMed Abstract | Crossref Full Text | Google Scholar
23. Park YW, Woo H, Jeong YY, Lee JH, Park JJ, Lee SS. Association of nodular regenerative hyperplasia of the liver with porto-pulmonary hypertension in a patient with systemic lupus erythematosus. Lupus. (2006) 15:686–8. doi: 10.1177/0961203306070976
PubMed Abstract | Crossref Full Text | Google Scholar
24. Suárez-Díaz S, García-Calonge M, Mendoza-Pacas G, Mozo-Avellaneda L, Caminal-Montero L. Non-cirrhotic portal hypertension in systemic lupus erythematosus. Cureus. (2023) 15:e35494. doi: 10.7759/cureus.35494
PubMed Abstract | Crossref Full Text | Google Scholar
25. Yamamoto M, Taniguchi H, Ohara M, Suzuki C, Naishiro Y, Ozeki I, et al. Beneficial effect of glucocorticosteroids for esophageal varices due to idiopathic portal hypertension following systemic lupus erythematosus. Nihon Rinsho Meneki Gakkai Kaishi. (2004) 27:40–7. doi: 10.2177/jsci.27.40
PubMed Abstract | Crossref Full Text | Google Scholar
26. Yang QB, He YL, Peng CM, Qing YF, He Q, Zhou JG. Systemic lupus erythematosus complicated by noncirrhotic portal hypertension: a case report and review of literature. World J Clin cases. (2018) 6:688–93. doi: 10.12998/wjcc.v6.i13.688
PubMed Abstract | Crossref Full Text | Google Scholar
27. Horita T, Tsutsumi A, Takeda T, Yasuda S, Takeuchi R, Amasaki Y, et al. Significance of magnetic resonance imaging in the diagnosis of nodular regenerative hyperplasia of the liver complicated with systemic lupus erythematosus: a case report and review of the literature. Lupus. (2002) 11:193–6. doi: 10.1191/0961203302lu164cr
PubMed Abstract | Crossref Full Text | Google Scholar
28. Inagaki H, Nonami T, Kawagoe T, Miwa T, Hosono J, Kurokawa T, et al. Idiopathic portal hypertension associated with systemic lupus erythematosus. J Gastroenterol. (2000) 35:235–9. doi: 10.1007/s005350050336
留言 (0)