
記住我
Creutzfeldt-Jakob disease (CJD) is a rare, fatal, rapidly progressive neurodegenerative disease resulting from an accumulation of misfolded prion proteins (PrP; Uttley et al., 2020; Maddox et al., 2020). Sporadic CJD (sCJD) is the most common form of CJD, comprising ~85–90% of cases (Uttley et al., 2020; Maddox et al., 2020). sCJD can be classified into subtypes based on the PrP type and polymorphism at codon 129 of the prion protein gene (PRNP) MM(MV)1, MV2, MM2, VV1, and VV2. sCJD typically affects middle-aged and elderly individuals with a median age at death of 67 years. Different subtypes have variable disease durations and phenotypes. MM1 and MV1 subtypes tend to present with cognitive decline with a fast progression (median: 3–4 months), while VV2 and MV2K subtypes present with cerebellar symptoms with longer disease duration (median: 6.5–17 months). MM2 and MV2C have longer disease duration with initial severe cognitive deficits (median 16 months; Collins et al., 2006). Idiopathic CJD in individuals with onset and death younger than 30 is very rare (Maddox et al., 2020). A few sCJD cases in younger individuals have been reported, with varying disease durations and phenotypes (Corato et al., 2006; Appleby et al., 2021; D'Arcy et al., 2019; Tam et al., 2023). Most young onset sCJD cases have been VV1, the rarest subtype, or sporadic fatal insomnia (sFI). It is extremely rare for a young sCJD patient to have the MV1 subtype: one case has been reported, which presented at 15-years-of-age and had a disease duration of nearly 10 years (D'Arcy et al., 2019). Herein, we describe a new case of MV1 sCJD with onset in the early 20's and a unusually long disease duration of 39 months especially for an MV1 subtype.
The patient was a 22-year-old Caucasian woman who initially presented to the emergency department with 7 month history of sudden neurocognitive decline with behavioral and personality changes. Her family history was significant only for late-onset dementia in her maternal grandmother. She had never received gonadotropin treatment, blood transfusions, or tissue transplants, and had never undergone neurosurgery. Her medical history was notable for depression and anxiety that began 3 years prior and had been stabilized with antidepressants, without any progression or additional neuropsychiatric symptoms. She had not traveled to bovine spongiform encephalopathy (BSE) affected countries during 1980–1996, nor did she have any history of venison consumption. She was not a hunter.
Her initial symptoms included new onset anxiety after she had returned home from college. Due to increased anxiety, she was seen by her primary care physician who placed the patient on scheduled escitalopram and clonazepam as needed. Despite the medication changes, her symptoms progressed.
Her anxiety continued in the following months, progressing to include symptoms of visual hallucinations, somnambulism, and insomnia. Three months after the initial onset of her symptoms, she started to demonstrate repetitive behaviors, including frequently repeating the same story and developing a ritualistic daily routine. She was insistent on washing her hands, flossing, and promptly brushing her teeth in this specific order, perseverating on this routine, and if not done in the correct order, she would forget steps. Shortly after these behavioral changes, she started to demonstrate more prominent cognitive deficits, having difficulty remembering the route to get home from her friend's house, the passcode on her phone, and even names of her immediate family members. She eventually developed difficulties in daily tasks, including putting on her shoes, often putting her left shoe on her right foot and vice versa. In addition, she demonstrated increased agitation and onset of ataxia. Previously a studious and well-adjusted college student, the patient's progressive cognitive decline and behavioral symptoms prompted her family to seek medical care.
On initial evaluation at the emergency department, the patient was noted to be avoidant and inattentive, with poor eye contact. She was alert and oriented to self and city, but not to date, including month or year. She scored a score of seven out of 30 on the Montreal Cognitive Assessment (MOCA), demonstrating deficits in short-term memory, recalling zero out of three objects, and inattentiveness, as evidenced by the inability to perform serial seven subtractions or count backward from 20.
On physical examination, she could not follow commands appropriately or perform finger-to-nose testing. Her pupils were equal, round, and reactive without neck stiffness or rigidity present. Dysarthria and aphasia were also absent. Equal and bilateral strength of upper and lower flexors and extensors were noted with +2/4 deep tendon reflexes of the biceps, brachioradialis, triceps, knee, and ankle. Unequivocal plantar reflexes were noted bilaterally with the Babinski maneuver.
Extensive testing was performed, including an autoimmune encephalitis and rheumatologic workup, which were negative. HIV and syphilis testing were negative as well, with TSH level and liver function tests within normal limits. The sedimentation rate was mildly elevated at 21 and CRP was within normal range. EEG demonstrated a normal study in wakefulness with no periodic sharp wave complexes (PSWC) or epileptiform activity. At clinical presentation, the initial brain MRI with and without contrast imaged at the first clinical presentation revealed diffuse bilateral cerebral cortical diffusion restriction with subtle sparing of the perirolandic cortex. No associated abnormal enhancement, abnormal meningeal enhancement, or mass was noted. Brain MRI at the initial evaluation revealed restricted diffusion in parietal, frontal, and occipital cortices bilaterally, consistent with CJD (Bizzi et al., 2021). Repeat MRI several months later demonstrated additional restricted diffusion in the insula, caudate, putamen, and thalami (Figures 1A, B). The Cerebrospinal fluid (CSF) analyses conducted at the National Prion Disease Pathology Surveillance Center (NPDPSC) 8 months following the onset showed elevated CSF 14-3-3 protein, total tau level of 4,471 ng/mL, and an indeterminate real-time quaking-induced conversion (RT-QuIC) result. Sequencing detected no pathogenic mutation in PRNP. She was heterozygous at the non-pathogenic polymorphism located on codon 129 (M129V).
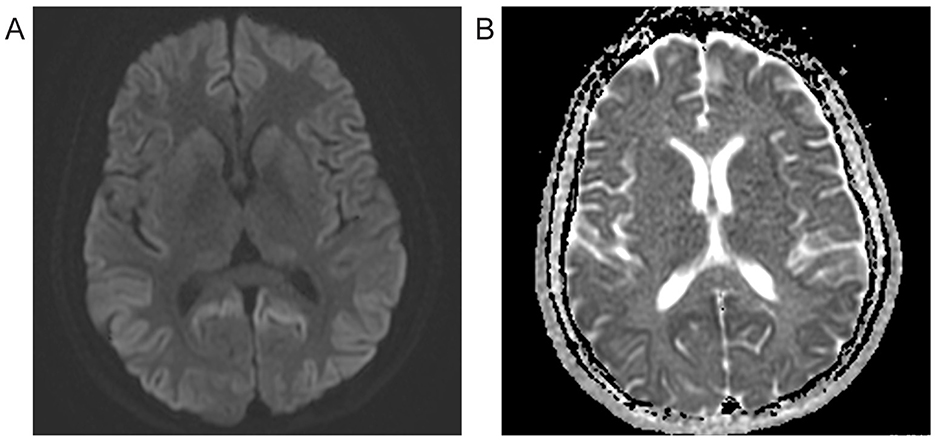
Figure 1. Brain MRI 8 months after symptom onset. (A) Diffusion-weighted imaging (DWI) reveals hyperintensity bilaterally in most cortices with additional restricted diffusion in the insula, caudate, putamen, and thalami. (B) Apparent diffusion coefficient (ADC) image shows hypointensity in hyperintense DWI sites, indicating restricted diffusion.
A few months after her emergency department visit, her cognition rapidly worsened with deterioration of language, executive, and visuospatial functions. She also developed delusions, headaches, severe gait ataxia with frequent falls, tremors, and hyperhidrosis, followed by myoclonus, insomnia, double incontinence, seizures, and visual dysfunction. Additionally, she developed new neuropsychiatric symptoms such as depression, anxiety, and extreme panic attacks that was no longer stabilized with antidepressants or antipsychotics. She did not undergo any life-prolonging interventions and died at home ~39 months from symptom onset (Table 1).
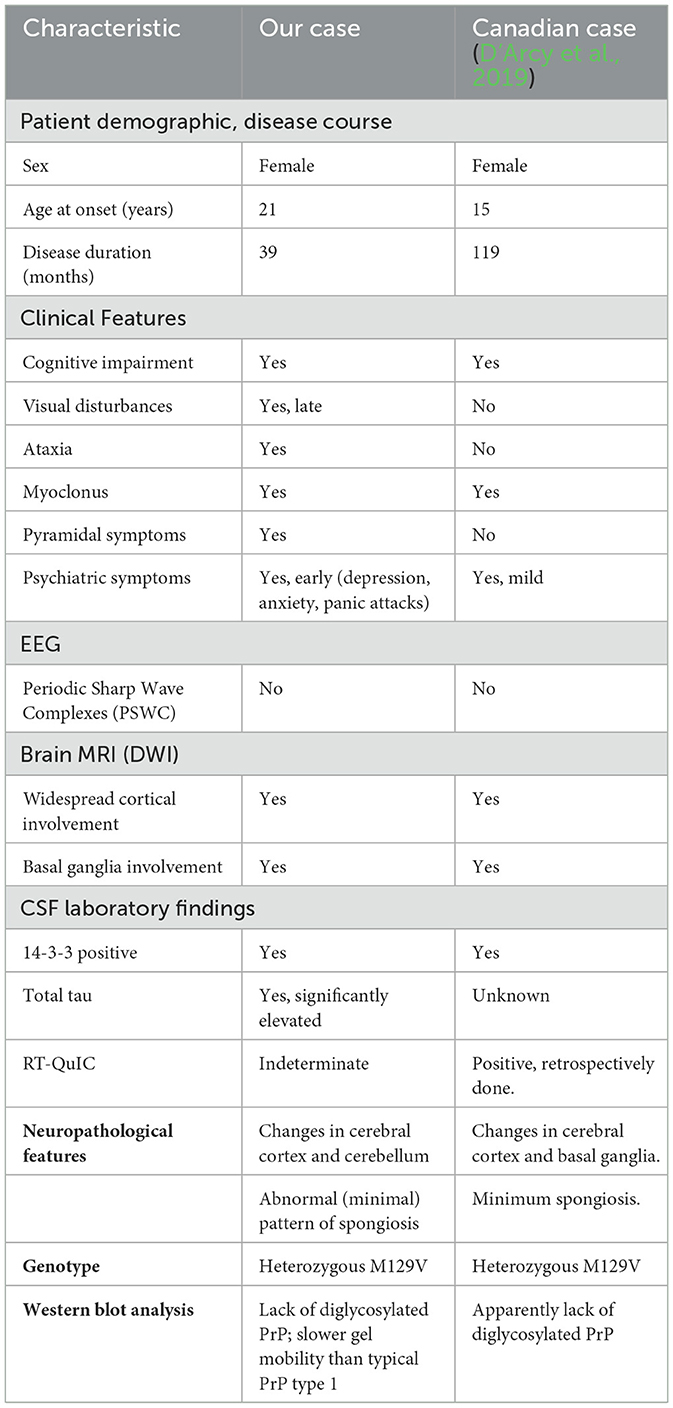
Table 1. Comparison of our case and the Canadian case from 2019.
Neuropathologic examination revealed a pale, thin neocortex with marked neuronal loss and gliosis (“status spongiosis;” Figures 2A–C). The hippocampus was relatively spared. Classical spongiform degeneration was not discernible (Figure 2). Immunoreactive PrP deposits were detected by immunohistochemistry throughout the neocortex and cerebellum (Figures 2D, E). No intracellular PrP deposits in Purkinje cells were observed (Figure 2E). Western blot analyses showed PK-resistant PrP bands at a slightly higher molecular weight than type 1 PrP associated with classic sCJD MM1, suggesting an atypical glycoform (AG) of PrP protein (sCJDMVAG; Figure 2F; Tanev and Yilma, 2009; Zanusso et al., 2007; Galeno et al., 2017).
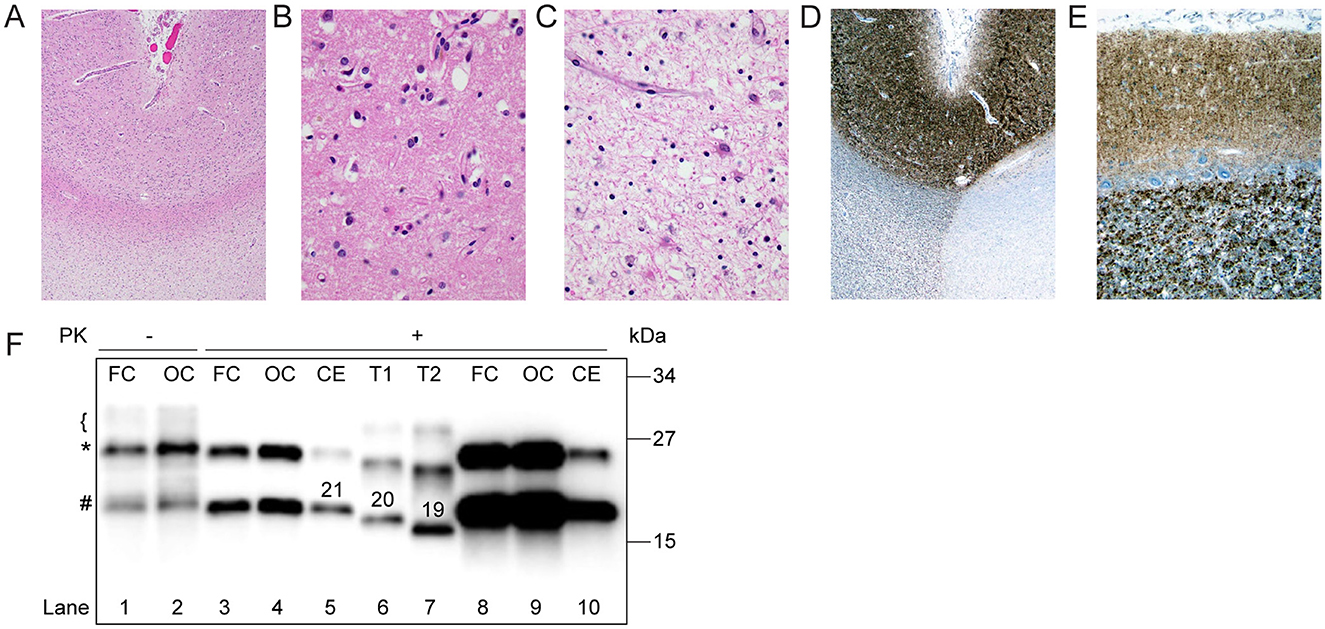
Figure 2. Histopathology and western blot profile of the index case. (A) Low-power H&E-stained section demonstrates pallor of subcortical white matter, consistent with secondary axonal degeneration (H and E x 20). (B) Cerebral cortical sections were remarkable for status spongiosis, rather than classical spongiform degeneration, secondary to advanced neuronal degeneration (H&E x 200). (C) Subcortical white matter was remarkable for severe axonal loss with reactive astrocytosis and scattered lipid-laden macrophages (H&E x 200). (D, E) 3 F4 immunohistochemical staining showed classical granular staining throughout the full-thickness of the cerebral cortex and cerebellar molecular layer. (F) Proteinase K (PK)-undigested PrPSc appears as a well-defined band of ~25 kDa (*), a less defined one of ~21 kDa (#), and a smear on the ~29–30 kDa region (bracket; lanes 1, 2). After digestion with PK, PrPSc is visualized as a doublet of ~ 25 and 21 kDa (lanes 3, 4, 8–10). Type 1 (T1, lane 6) and type 2 (T2, lane 7) controls were obtained from sCJDMM1 and -MM2, respectively. Numbers atop PrPSc bands indicate the relative molecular mass. FC, Frontal cortex; OC, occipital cortex; CE, cerebellum. Loading volume of lanes 8 and 9 is 4-fold that of lanes 3 and 4, respectively.
DiscussionVery young decedents (before the age of 30) with CJD are extremely rare, and most are attributable to exogenous factors or inherited genetic mutation. Only a handful have sCJD, and these are usually either sCJDVV1 subtypes or sporadic fatal insomnia (sFI) that typically have a younger disease onset (40–50's) and a longer disease duration than other subtypes (Maddox et al., 2020; Abu-Rumeileh et al., 2018; Brown et al., 1984; Cali et al., 2020). Until recently, young onset has not been reported in classical sCJD MV1 cases or sCJD MV1 cases with atypical histopathological phenotypes (Gelpi et al., 2022; Cracco et al., 2023). The first very young-onset sCJD MV1 subtype with an unexpectedly long disease duration (119 months, which may be in part due to life-prolonging interventions) was recently reported in Canada (D'Arcy et al., 2019). Our patient, who developed her first symptoms of depression and anxiety at 21 years of age, is the second sCJDMV1 case, to our knowledge, with a notably young disease onset. The longer duration of illness seen in both cases may be attributable to young age since younger patients are less likely to have premorbid neurological conditions and comorbidities. Reported young onset sCJDs (before the age of 50) patients have, on average 1.45 months longer disease duration (Corato et al., 2006). However, in the NPDPSC's cohort of 168 sCJDMV1 cases, the mean age (67 years) of the patients with disease duration ranging 1–10 months (N = 137; median: 3 months) does not differ from that (68 years) of cases with a duration ranging 11–41 months (N = 31; median: 16 months). These data suggest that the longer survival seen in typical sCJDMV1 cases is not simply due to the young age of onset.
Classically, the median age for the sCJD MV1 subtype is 65.5 years, with a median disease duration of 5 months (Collins et al., 2006). sCJD MV1 subtype patients most commonly present with cognitive impairment such as memory loss, confusion, or disorientation (Collins et al., 2006; Gelpi et al., 2022). Certain atypical characteristics such as neuropsychiatric symptoms, headaches, and sleep disturbances are more commonly reported in young onset sCJDs (Corato et al., 2006; Appleby et al., 2007). Both the Canadian case and our case presented like most young sCJD patients who initially present with neuropsychiatric symptoms and then develop cognitive symptoms, culminating in gross neurological deficits (D'Arcy et al., 2019; Cracco et al., 2023).
Furthermore, our case characterized by underrepresentation of the diglycosylated PrP isoform (Figure 2F; D'Arcy et al., 2019). Despite the young age, the lack or marked underrepresentation of PK-resistant diglycosylated PrP argue against the diagnosis of variant CJD (Diack et al., 2019). Our patient initially presented with neuropsychiatric symptoms like depression and anxiety followed by signs of diffuse cognitive impairments such as memory loss, delusions, and headaches. Her neuropsychiatric symptoms early during her disease course could either be idiopathic or an early presentation of sCJDs (Corato et al., 2006; Appleby et al., 2007). However, given that the patient's mood was previously well-controlled with antidepressants and that she developed new and progressively worsening symptoms of depression, anxiety, and panic attacks, we attribute the latter symptoms to her new onset sCJD. The patient progressively developed worsening diffuse cognitive impairment, motor symptoms, and myoclonus. Despite the differences in the survival time between the Canadian case (119 months) and our case (39 months), the clinical progression of the two cases were similar (Table 1).
PSWC are seen in most sCJD MV1 subtype patients (Parchi et al., 1999); however, this finding is often time-dependent and may be missed in individual cases. Interestingly, neither the Canadian nor our case had the characteristic PSWCs (D'Arcy et al., 2019). Both were positive for the cerebrospinal fluid (CSF) level of the 14-3-3 protein, a surrogate marker of CJD (Castellani et al., 2004; Otto et al., 2002). RT-QuIC is a relatively new diagnostic method with high sensitivity and specificity that utilizes the PrP protein's self-replicating ability and aggregate formation (Green, 2019; Rhoads et al., 2020). The Canadian case tested positive for RT-QuIC, but our case was deemed indeterminate due to the assay's low amplitude that did not reach positivity threshold. In our case, a long lag time was also noted on CSF RT-QuIC. Early MRI images from both cases revealed hyperintensity bilaterally in most cortices, consistent with characteristic MV1 subtype images (Gelpi et al., 2022). Later, MRI images showed additional diffusion in the striatum in both cases (Figure 1).
Neuropathological examination of sCJD MV1 subtype patients reveals spongiform changes in most layers of the cortex, sparing the first layer, astrogliosis, and neuronal loss. The molecular layer is primarily affected in the cerebellum, and the hippocampus is usually spared (Parchi et al., 1999). Neuropathologic analyses of our case showed generalized cortical atrophy and signs of astrogliosis but had minimal spongiform changes. The Canadian case also had minimal spongiform changes. Immunohistochemical staining revealed a dense deposition of PrP proteins in the neocortex and cerebellum, confirming the diagnosis (Figures 2A–E). Interestingly, the western immunoblot analysis for protease-resistant PrP showed PK-resistant PrP bands at slightly higher molecular weight than type 1 PrP, suggesting an atypical glycoform of PrP protein (Figure 2F).
ConclusionIn summary, very young sCJD patients with long disease duration may present quite differently than typical sCJD patients. To our knowledge, our case is the second reported case of a very young onset sCJD MV1 subtype with an unusually long disease duration and PrP type. Our case's MRI findings are consistent with what we would expect from an MV1 subtype patient. However, the clinical presentation, disease course, the absence of PSWCs in EEG, and indeterminate RT-QuIC results is not characteristic of sCJD MV1. Ultimately, the neuropathological exam analyses performed at the NPDPSC combined with no known risk factors for acquired prion disease provided confidence that this young patient had sporadic prion disease. An extremely rare, very young onset sCJD with a long disease duration case may present differently from the characteristic clinical presentations of sCJD. Thus, it is critical to comprehensively evaluate clinical findings, but most importantly to verify the presence and type of prion disease in these younger cases though neuropathologic evaluation at autopsy, especially since acquired prion diseases historically occur in young individuals (Rhoads et al., 2020; Manix et al., 2015). Neuropathologic evaluation of young cases in the UK was crucial for identifying variant CJD in those exposed to bovine spongiform encephalopathy (Will et al., 1996). The U.S. is currently dealing with a massive spread of an animal prion disease in the cervid species called chronic wasting disease. As such, it is more important than ever to confirm the presence and type of prion disease is the US, especially in atypical cases of prion disease (Osterholm et al., 2019).
Data availability statementThe datasets presented in this article are not readily available because, the dataset is not accessible to the general public due to privacy concerns. Requests to access the datasets should be directed to Brian Stephen Appleby, YnNhMzUmI3gwMDA0MDtjYXNlLmVkdQ==.
Ethics statementThe studies involving humans were approved by University Hospitals Cleveland Medical Center IRB. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributionsLA: Writing – original draft, Conceptualization, Writing – review & editing. MC: Formal analysis, Supervision, Writing – review & editing. IC: Data curation, Investigation, Writing – review & editing. TR: Formal analysis, Writing – review & editing. JL: Formal analysis, Writing – review & editing. XJ: Data curation, Writing – review & editing, Formal analysis. AB: Writing – review & editing. LS: Writing – review & editing. RM: Writing – review & editing. RP: Writing – review & editing, Data curation. TG: Writing – review & editing, Data curation. JG: Writing – review & editing. MH: Writing – review & editing. BA: Conceptualization, Formal analysis, Funding acquisition, Investigation, Project administration, Supervision, Writing – review & editing.
FundingThe author(s) declare financial support was received for the research, authorship, and/or publication of this article. The National Prion Disease Pathology Surveillance Center was awarded NU38CK000486 from the CDC for continuing an enhanced and multifaceted national surveillance program for human prion diseases.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statementThe author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher's noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimerThe findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
ReferencesAbu-Rumeileh, S., Redaelli, V., Baiardi, S., Mackenzie, G., Windl, O., Ritchie, D. L., et al. (2018). Sporadic fatal insomnia in Europe: phenotypic features and diagnostic challenges. Ann. Neurol. 84, 347–360. doi: 10.1002/ana.25300
PubMed Abstract | Crossref Full Text | Google Scholar
Appleby, B. S., Appleby, K. K., and Rabins, P. V. (2007). Does the presentation of Creutzfeldt-Jakob disease vary by age or presumed etiology? a meta-analysis of the past 10 years. J. Neuropsychiat. Clin. Neurosci. 19, 428–435. doi: 10.1176/jnp.2007.19.4.428
PubMed Abstract | Crossref Full Text | Google Scholar
Appleby, B. S., Maddox, R., Schonberger, L. B., Cali, I., Hammett, T., Cohen, M., et al. (2021). Sporadic Creutzfeldt-Jakob disease in a very young person. Neurology 97, 813–816. doi: 10.1212/WNL.0000000000012737
PubMed Abstract | Crossref Full Text | Google Scholar
Bizzi, A., Pascuzzo, R., Blevins, J., Moscatelli, M. E. M., Grisoli, M., Lodi, R., et al. (2021). Subtype diagnosis of sporadic Creutzfeldt-Jakob disease with diffusion magnetic resonance imaging. Ann. Neurol. 89, 560–572. doi: 10.1002/ana.25983
PubMed Abstract | Crossref Full Text | Google Scholar
Brown, P., Rodgers-Johnson, P., Cathala, F., Gibbs, C. J., and Gajdusek, D. C. (1984). Creutzfeldt-Jakob disease of long duration: clinicopathological characteristics, transmissibility, and differential diagnosis. Ann. Neurol. 16, 295–304. doi: 10.1002/ana.410160305
PubMed Abstract | Crossref Full Text | Google Scholar
Cali, I., Puoti, G., Smucny, J., Curtiss, P. M., Cracco, L., Kitamoto, T., et al. (2020). Co-existence of PrPD types 1 and 2 in sporadic Creutzfeldt-Jakob disease of the VV subgroup: phenotypic and prion protein characteristics. Sci. Rep. 10:1503. doi: 10.1038/s41598-020-58446-0
PubMed Abstract | Crossref Full Text | Google Scholar
Castellani, R. J., Colucci, M., Xie, Z., Zou, W., Li, C., Parchi, P., et al. (2004). Sensitivity of 14-3-3 protein test varies in subtypes of sporadic Creutzfeldt-Jakob disease. Neurology 63, 436–442. doi: 10.1212/01.WNL.0000135153.96325.3B
PubMed Abstract | Crossref Full Text | Google Scholar
Collins, S. J., Sanchez-Juan, P., Masters, C. L., Klug, G. M., van Duijn, C., Poleggi, A., et al. (2006). Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain 129, 2278–2287. doi: 10.1093/brain/awl159
PubMed Abstract | Crossref Full Text | Google Scholar
Corato, M., Cereda, C., Cova, E., Ferrarese, C., and Ceroni, M. (2006). Young-onset CJD: age and disease phenotype in variant and sporadic forms. Funct. Neurol. 21, 211–215.
PubMed Abstract | Google Scholar
Cracco, L., Puoti, G., Cornacchia, A., Glisic, K., Lee, S. K., Wang, Z., et al. (2023). Novel histotypes of sporadic Creutzfeldt-Jakob disease linked to 129MV genotype. Acta Neuropathol. Commun. 11:141. doi: 10.1186/s40478-023-01631-9
PubMed Abstract | Crossref Full Text | Google Scholar
D'Arcy, C. E., Bitnun, A., Coulthart, M. B., D'Amour, R., Friedman, J., Knox, J. D., et al. (2019). Sporadic Creutzfeldt-Jakob disease in a young girl with unusually long survival. J. Neuropathol. Exp. Neurol. 78, 373–378. doi: 10.1093/jnen/nlz013
PubMed Abstract | Crossref Full Text | Google Scholar
Diack, A. B., Boyle, A., Plinston, C., Hunt, E., Bishop, M. T., Will, R. G., et al. (2019). Variant Creutzfeldt-Jakob disease strain is identical in individuals of two PRNP codon 129 genotypes. Brain 142:awz076. doi: 10.1093/brain/awz076
PubMed Abstract | Crossref Full Text | Google Scholar
Galeno, R., Bari, M. A. D., Nonno, R., Cardone, F., Sbriccoli, M., Graziano, S., et al. (2017). Prion strain characterization of a novel subtype of Creutzfeldt-Jakob disease. J. Virol. 91:e02390–16. doi: 10.1128/JVI.02390-16
PubMed Abstract | Crossref Full Text | Google Scholar
Gelpi, E., Baiardi, S., Nos, C., Dellavalle, S., Aldecoa, I., Ruiz-Garcia, R., et al. (2022). Sporadic Creutzfeldt-Jakob disease VM1: phenotypic and molecular characterization of a novel subtype of human prion disease. Acta Neuropathol. Commun. 10:114. doi: 10.1186/s40478-022-01415-7
PubMed Abstract | Crossref Full Text | Google Scholar
Maddox, R. A., Person, M. K., Blevins, J. E., Abrams, J. Y., Appleby, B. S., Schonberger, L. B., et al. (2020). Prion disease incidence in the United States: 2003–2015. Neurology 94, e153–e157. doi: 10.1212/WNL.0000000000008680
PubMed Abstract | Crossref Full Text | Google Scholar
Manix, M., Kalakoti, P., Henry, M., Thakur, J., Menger, R., Guthikonda, B., et al. (2015). Creutzfeldt-Jakob disease: updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy. Neurosurg. Focus. 39:E2. doi: 10.3171/2015.8.FOCUS15328
PubMed Abstract | Crossref Full Text | Google Scholar
Osterholm, M. T., Anderson, C. J., Zabel, M. D., Scheftel, J. M., Moore, K. A., Appleby, B. S., et al. (2019). Chronic wasting disease in cervids: implications for prion transmission to humans and other animal species. mBio 10, e01091–e01019. doi: 10.1128/mBio.01091-19
PubMed Abstract | Crossref Full Text | Google Scholar
Otto, M., Wiltfang, J., Cepek, L., Neumann, M., Mollenhauer, B., Steinacker, P., et al. (2002). Tau protein and 14-3-3 protein in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology 58, 192–197. doi: 10.1212/WNL.58.2.192
PubMed Abstract | Crossref Full Text | Google Scholar
Parchi, P., Giese, A., Capellari, S., Brown, P., Schulz-Schaeffer, W., Windl, O., et al. (1999). Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann. Neurol. 46, 224–233. doi: 10.1002/1531-8249(199908)46:2<224::AID-ANA12>3.0.CO;2-W
Crossref Full Text | Google Scholar
Rhoads, D. D., Wrona, A., Foutz, A., Blevins, J., Glisic, K., Person, M., et al. (2020). Diagnosis of prion diseases by RT-QuIC results in improved surveillance. Neurology 95, e1017–e1026. doi: 10.1212/WNL.0000000000010086
PubMed Abstract | Crossref Full Text | Google Scholar
Tam, J., Centola, J., Kurudzhu, H., Watson, N., MacKenzie, J., Leitch, M., et al. (2023). Sporadic Creutzfeldt-Jakob Disease in the young (50 and below): 10-year review of United Kingdom surveillance. J. Neurol. 270, 1036–1046. doi: 10.1007/s00415-022-11467-3
PubMed Abstract | Crossref Full Text | Google Scholar
Uttley, L., Carroll, C., Wong, R., Hilton, D. A., and Stevenson, M. (2020). Creutzfeldt-Jakob disease: a systematic review of global incidence, prevalence, infectivity, and incubation. Lancet Infect. Dis. 20, e2–e10. doi: 10.1016/S1473-3099(19)30615-2
PubMed Abstract | Crossref Full Text | Google Scholar
Will, R. G., Ironside, J. W., Zeidler, M., Cousens, S. N., Estibeiro, K., Alperovitch, A., et al. (1996). A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 347, 921–925. doi: 10.1016/S0140-6736(96)91412-9
PubMed Abstract | Crossref Full Text | Google Scholar
Zanusso, G., Polo, A., Farinazzo, A., Nonno, R., Cardone, F., Di Bari, M., et al. (2007). Novel prion protein conformation and glycotype in Creutzfeldt-Jakob disease. Arch. Neurol. 64, 595–599. doi: 10.1001/archneur.64.4.595
留言 (0)