
記住我

To investigate intermolecular interactions between peptides and polyphenols, six homopolypeptides containing different side chains (i.e., Arg6, His6, Lys6, Asp6, Pro6, and Thr6) were chosen as model peptides, while TA was selected as the model polyphenol (Scheme 1a). Notably, the amino group in the peptide’s amino acid is easily protonated and positively charged, while the carboxyl group is easily deprotonated and negatively charged, depending on the isoelectric point of the amino acid and solution pH [29]. Similarly, the TA phenolic hydroxyl group is also easily deprotonated and negatively charged under acidic conditions [30], which provides preconditions for electrostatic interactions between polyphenols and polypeptides. In addition, the selected peptides can form other noncovalent interactions, such as hydrogen-bonding, or hydrophobic interactions with TA. Subsequently, polyphenol–protein coatings were obtained by dip-coating and LbL assembly on silicon substrates, and the assembly mechanism was investigated (Scheme 1b).
Scheme 1
Schematics of (a) (i) peptide and (ii) TA structures and (b) experimental design and LbL assembly
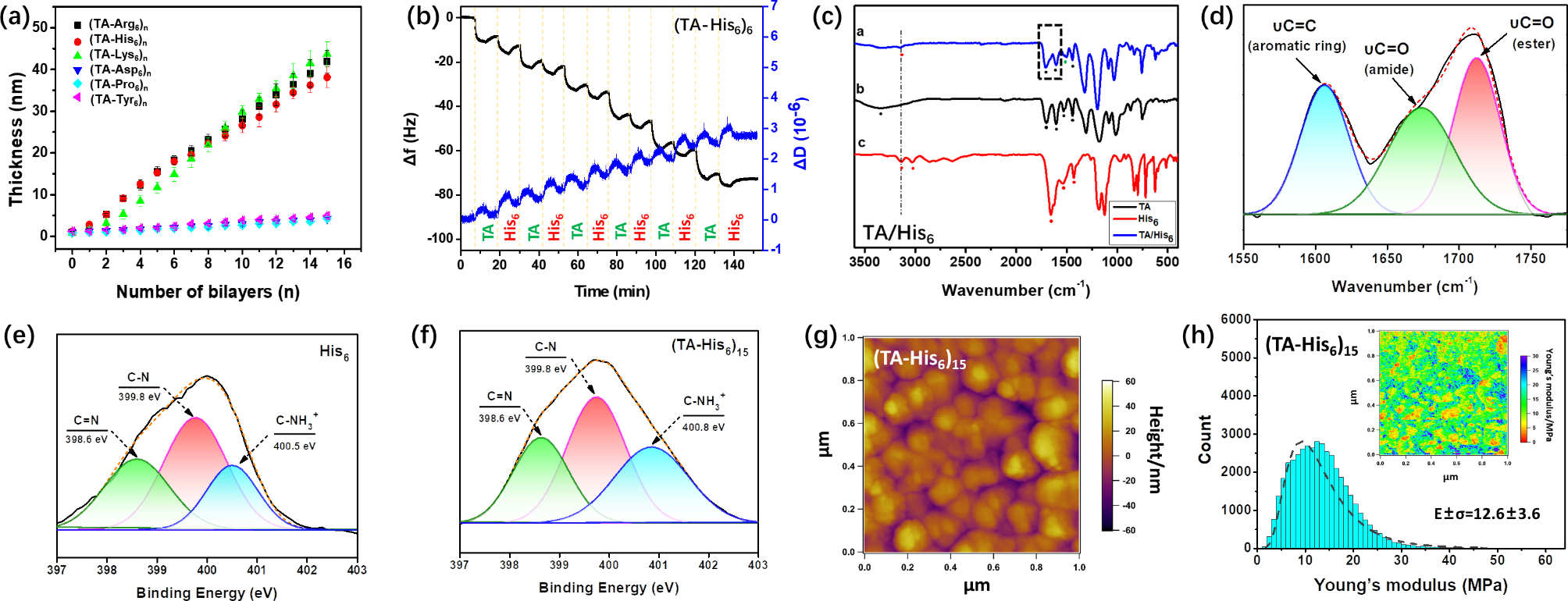
LbL assemblyFigure 1a shows the thickness growth of hexapeptides (hexameric amino acids) assembled with positively and negatively charged, uncharged, and hydrophobic TA layers. First, the thicknesses of the layered negatively charged peptide (Asp6), uncharged peptide (Pro6), and hydrophobic peptide (Tyr6) assembled with TA negligibly increased, while the thicknesses of the layered coatings of positively charged peptides Arg6, His6, and Lys6 assembled with TA all significantly increased, and the thickness of 15 bilayers was similar, approximately 40 nm, which indicates that the TA–peptide assembly was dominated by electrostatic interactions, while neither hydrogen-bonding nor hydrophobic interactions alone maintained the LbL assembly of TA with peptides. Figure 1b shows the frequency (f) plotted as a function of the dissipation (D) during the assembly of TA with the positively charged peptide (His6) at pH 7, where f is inversely proportional to the sensor-coupling quality, and D is a semiquantitative parameter to express the “softness” of films [31]. During the LbL assembly of the TA and positively charged peptide, Δf monotonically decreased, indicating that the TA and peptide were both deposited on the substrate. Notably, Δf was significantly higher for the TA layer than the peptide, indicating that during the LbL assembly, more TA was deposited than the peptide. Additionally, ΔD monotonically increased during both the TA and peptide assemblies, indicating that the coating became softer and swollen with increasing number of assembled layers. For both the TA and His6 layers, ΔD was low and similar, indicating that the TA and His6 layers were structurally similar and that the coatings exhibited relatively rigid mechanical properties. In addition, during the assemblies of TA with Arg6 (Figure S1a) and Lys6 (Figure S1b), Δf and ΔD showed patterns consistent with that during the assembly of TA with His6.
Figure 1c shows the FTIR spectra for coatings of TA assembled with the positively charged peptide (His6) at pH 7. For TA, the broad peak at 3350 cm− 1 corresponds to the stretching vibration of ph-OH. The peak at 1701 cm− 1 corresponds to C = O stretching, and peaks at 1605, 1533, and 1443 cm− 1 correspond to skeletal vibrations of the benzene ring. For the His6 peptide, moderately intense peaks at 3137 and 3031 cm− 1 are both antisymmetric stretching vibrations of N–H in the amide. The peak at 1657 cm− 1 corresponds to the stretching vibration of the amide I band (C = O). The peak at 1543 cm− 1 corresponds to the bending vibration of the amide II band (N–H). The peak at 1430 cm− 1 corresponds to the absorption of the amide III band (C–N). For TA–His6, the peak at 3152 cm− 1 corresponds to the stretching vibration of the amide N–H of His6. The peak at 1710 cm− 1 corresponds to the stretching vibration of the ester carbonyl C = O of TA. The peak at 1674 cm− 1 corresponds to the stretching vibration of the amide I band of His6 (Fig. 1d). Peaks at 1605, 1503, 1444 cm− 1 correspond to skeletal vibrations of the benzene ring in TA, where the peak at 1503 cm− 1 indicates that some phenolic hydroxyl groups in TA were oxidized to quinone structures, and the peak at 1444 cm− 1 corresponds to the absorption of the amide III band (C–N) of His6. These results suggest that TA was assembled with His6. Similar FTIR spectra were obtained for the assemblies of TA with Arg6 (Figure S2a) and Lys6 (Figure S2b), indicating that TA was assembled with positively charged peptides.
Due to the high oxidative susceptibility of TA, especially when pH > 7, the phenolic hydroxyl group can form reversible covalent Schiff-base interactions with amines after oxidation to quinone (C = N). [32] His6 was characterized using XPS before (Fig. 1e) and after (Fig. 1f) LbL assembly with TA, and the N 1 s peak was deconvoluted in the His6 spectrum but not in the TA spectrum. The C = N content was negligibly different before and after the LbL assembly, indicating that the coating contained almost no Schiff-base covalent bonds. Similar XPS spectra were obtained for the assemblies of TA with Arg6 (Figure S3a and b) and Lys6 (Figure S3c and d), and the N 1 s compositions quantitatively analyzed before and after the assembly are listed in Table S3. The absence of Schiff base in the LbL coating is partly due to the slower oxidation of TA under neutral conditions and the formation of less quinone, and partly due to the short assembly time is not sufficient for the formation of Schiff base covalent bonds. In addition, the coating compositions of TA assembled with different positively charged peptides were analyzed using XPS (Table S4). For Arg6, His6, and Lys6, the proportion of bound TA decreased. This may be due to decreases in the number of protonatable N atoms and the density of positive charges on the peptide surface, which weakened electrostatic interactions and, thus, decreased the amount of bound TA.
Because properties, such as morphology and mechanical properties, affect biological functions of peptide coatings under physiological conditions, LP–AFM tests were conducted by immersing peptide coatings in buffer solutions. LP–AFM enables the evaluation of the structure and properties of biologically active coatings under physiological conditions while reducing the amount of damage to the product [33]. Figure 1g shows the surface morphology of the (TA–His6)15 coating in the liquid environment. The coating consists of aggregated particles with a discontinuous rough surface structure, and the average height of the coating is approximately 40 nm, which is consistent with the optical thickness measured using ellipsometric polarization spectroscopy. The Young’s modulus of the (TA–His6)15 coating was further evaluated. As shown in Fig. 1h, the mechanical properties of the coating were relatively uniformly distributed, and a Young’s modulus of approximately 12.6 ± 3.6 MPa showed a Gaussian distribution. Coatings (TA–Arg6)15 and (TA–Lys6)15 also exhibited morphologies and Young’s modulus distributions similar to those of (TA–His6)15 (Figs. S4 and S5, respectively). Young’s modulus was obtained by fitting the LP–AFM force curve to the Johnson–Kendall–Roberts (JKR) model, which is a contact mechanics model that considers adhesive interactions in the contact zone [34]. Typical evaluated force curves coincided well with the JKR model curves, indicating the reliability of the Young’s modulus calculations (Figure S6). The surface adhesion work of the peptide coatings was then solved using the JKR theoretical equation, which indicates the coating adhesions [35], and the adhesion works of the (TA–Arg6)15, (TA–His6)15, and (TA–Lys6)15 coatings were 2.7, 4.1, and 3.6 µN·m− 1, respectively (Figure S7). In addition, the surface morphology, Young’s modulus, and adhesion work of the peptide coatings were evaluated in ambient air (Figure S8 and S9). Although the thicknesses of the peptide coatings obtained in the ambient air and liquid were negligibly different, the Young’s modulus and adhesion work of the peptide coatings obtained in ambient air were higher than those of the peptide coatings obtained in the liquid, which might be because of the difference in the film hydration in different testing environments [26].
Fig. 1
(a) Buildup curve for thickness of (TA/hexapeptide)n coating. (b) Real-time shifts of Δf and ΔD during fabrication of (TA/His6)6 coatings. (c) FTIR spectra for chemical structures of TA/His6, TA, and His6. (d) Detailed FTIR spectrum corresponding to area in dashed box in (c) for TA/His6. Detailed N 1 s XPS spectra of (e) His6 powder and (f) (TA–His6)15 coating on silicon wafers. (g) LP–AFM image of (TA–His6)15 coating. (h) Image and frequency distribution of Young’s modulus for (TA–His6)15 coating obtained in liquid
Influencing factors of LbL assemblyThe effects of the polyphenol and polypeptide species on the LbL assembly were investigated. Polyphenols having different molecular weights (TA, PC, EGCG, Cat, and GA, for which structures are displayed in Figure S10) were assembled with peptides. As shown in Fig. 2a, none of the polyphenols could be assembled with negatively charged (Asp6), uncharged (Pro6), and hydrophobic (Tyr6) peptides. TA, PC, EGCG, which had relatively high molecular weights, could be assembled with positively charged peptides (Arg6, His6, and Lys6), while Cat could only be assembled with Arg6, and GA, which had the lowest molecular weight, could not assemble with positively charged peptides, which indicates that the assembly of polyphenols and peptides mainly depends on electrostatic interactions and that the molecular weight affects the ability of polyphenols to assemble with peptides. In addition, Arg has slightly more protonatable N atoms than His and Lys and, thus, can form stronger electrostatic interactions with polyphenols.
The effects of the pH, salt concentration, and peptide chain length and type on the thickness of TA–peptide coatings were investigated during LbL assembly. For the effect of the pH on the thickness of TA–peptide coatings during LbL assembly, as shown in Fig. 2b, the assembly of TA with the positively charged peptide (His6) shows an obvious pH dependence, which is closely related to the pKa of TA, isoelectric point (pl), and number of charged amino acids in the peptide. The TA presents two pKa values, pKa1 = 3.3 and pKa2 = 8.7, and the pl of His is 7.6 [36,37,38]. With increasing pH, the thickness of the coating in which TA was assembled with His6 first increased and then decreased. When the solution is too acidic (pH < 3), the TA dissociation is inhibited, and the TA charge is reduced, which weakens the electrostatic interaction between TA and His6, resulting in almost no assembly growth of the coating. When the solution pH is above the pl of His6 (pH > 7.6), His6 N atoms protonate, and the His6 charge decreases, which weakens the electrostatic interaction between TA and His6 and slows the growth of the coating. In addition, for positively charged peptides Arg6 (pl = 10.8, Figure S11a) [39] and Lys6 (pl = 9.7, Figure S11b) [40], the pH dependence of the assemblies with TA showed patterns consistent with the pH dependence of the assembly of His6 with TA, indicating the dominance of electrostatic interactions in the LbL assembly of TA with peptides. The effect of the salt concentration on the coating thickness was investigated during the assembly of TA with positively charged peptides, as shown in Fig. 2c and Figure S12. LbL assembly negligibly depended on the salt concentration, which may be because the molecular weights of the TA and peptides are too low for the salt to effectively shield charges carried by these small molecules, thus rendering LbL assembly insensitive to the salt concentration.
The effect of the peptide chain length on the growth of the assembled coating was investigated by varying the number of amino acids in the peptide. His3, His6, His9, and His12 were designed and synthesized using the His unit. As shown in Fig. 2d, His3 could not effectively maintain the growth of coatings assembled LbL with TA, and the coating thickness significantly increased with increasing chain length. The thickest coating was prepared using His9. This might be because the number of charges carried by the positively charged peptide increased with increasing chain length, which enhanced the interaction between the TA and peptide and, thus, increased the film thickness with increasing chain length. However, the coating in which His12 was assembled with TA was slightly thinner than that in which His9 was assembled with TA. This might be because when the chain is too long, the potential resistance between the chain structures might weaken the interaction between the TA and peptide, which decreases the film thickness.
The effect of the peptide species on the thickness of the peptide coating was investigated by replacing the amino-acid species in the positively charged peptide chain. As shown in Fig. 2e and f, the positively charged peptide (Lys6) was used as a template, and two positively charged Lys amino acids were replaced with two negatively charged Asp, uncharged Gly, or hydrophobic Ile or Phe amino acids and assembled with TA. Clearly, the coating comprising Lys6 assembled with TA was the thickest, and the coating in which Lys4–Asp2 was assembled with TA grew the slowest. This is because the charge densities of all the peptides that were substituted with negatively charged and uncharged and hydrophobic amino acids were reduced to different degrees compared with the original positively charged peptide (Lys6). In particular, the deprotonated substitution of negatively charged amino acids cancels the protonation-induced positive charge in the basic negatively charged chain, which substantially reduces the positive charge density on the peptide chain surface and, thus, slows the growth of the LbL-assembled coating. Although uncharged amino-acid substitution may somewhat enhance the hydrogen-bonding interaction of the peptide with TA [41], the assembly of the peptide with TA is still much slower than that of the peptide with unsubstituted Lys6, indicating that hydrogen-bonding interactions play a lesser role than electrostatic interactions for assembling TA with peptides. Although hydrophobic amino-acid substitution could somewhat improve the hydrophobic interaction between the peptide and TA, the assembly of the peptide with TA was still somewhat slower, indicating that hydrophobic interactions play a lesser role than electrostatic interactions for assembling TA with peptides. Therefore, for assembling TA with peptides LbL, electrostatic interactions are dominant. Notably, peptide coatings prepared using polyphenol building blocks show good substrate universality because polyphenols readily adsorb on various material substrates [42]. As shown in Fig. 2g, polypeptide coatings were prepared on Ti, Si, PS, quartz, and glass substrates, and the substrate color obviously changed from light blue to light yellow before and after coating, respectively, indicating that coatings were successful deposited, where light yellow originates from TA.
Fig. 2
(a) Assembly of different polyphenols with peptides (smiling and crying emoticons mean can and cannot be assembled, respectively). Effects of (b) pH, (c) salt concentration, (d) peptide chain length, and (f) amino-acid types on coating growth. (e) Schematic showing substitution of two amino acids in Lys6 peptide. (g) Digital photographs of (TA–Lys6)15 coatings fabricated on Ti, Si, PS, quartz, and glass substrates
Stability is an important factor for biological applications of coatings [43]. DMEM contains various amino acids and glucose and is widely used for culturing various cells [44]. Coating stability under physiological conditions can be understood by observing changes in the thickness of coatings immersed in DMEM. As shown in Figure S13a, the coating thickness negligibly decreased with extended immersion in DMEM until the 7th day, indicating the good stability of the coating under physiological conditions. Additionally, the coating did not degrade when immersed in low (Figure S13b) and high (Figure S13c) concentrations of NaCl solutions, indicating that the coating has good salt resistance. In addition, the coating also exhibits good urea resistance (Figure S13d). The good stability of the coating is attributed partly to the good charge-matching effect between the positively charged peptide and the negatively charged polyphenol, but also to the stronger interaction force provided by the small-molecule assembly between the short peptide and the small-molecule polyphenol. Notably, by separately immersing the as-prepared peptide coatings in different pH buffers, the coatings had significant pH-responsive properties. Although the coating shows good stability under mildly acidic conditions (pH 5, Figure S13e), it rapidly decomposes under strongly acidic conditions (pH < 3, Figure S13f). Therefore, the TA–peptide coating has good potential for application to acid-responsive drug carriers.
Thermodynamic mechanisms of TA and peptide assemblyIsothermal titration calorimetry was used to determine the thermodynamics of interactions between the peptide and TA and obtain binding thermodynamic parameters, such as enthalpy, entropy, and Gibbs free-energy changes (ΔH, ΔS, and ΔG, respectively). For these measurements, TA solutions were loaded into the injection syringe and titrated into different peptide solutions in sample cells. The enthalpy change was monitored (Figure S14) and then plotted as a function of the molar ratio of the peptide and TA (Fig. 3a–g). The experimentally measured titration enthalpy of the TA and peptide was obtained by subtracting the dilution enthalpy of the buffer–TA (Figure S14h) from that of the pulse signal for the TA solution injected into the peptide solution (Figure S14 a–g). All the evaluated peptides that complexed with TA showed a significant change in enthalpy, indicating interaction and binding between the TA and peptide. The endpoint exotherms of the titration between TA and different peptides were approximately 0, and all the titration exotherms showed a good fit with the analytical model (Fig. 3a–g), indicating saturated binding between molecules and plausible data results. The fitted thermodynamic parameters (ΔH, ΔG, and − TΔS) obtained by titrating TA with different peptides are listed in Fig. 3h. All the evaluated peptides that complexed with TA showed a negative ΔG, indicating that the association was spontaneous. However, ΔH and − TΔS were substantially different for the TA and different peptides, indicating different driving forces and interactions. Binding between positively charged peptides (Arg6, His6, and Lys6) and TA was predominantly enthalpy driven with a high ΔH and an unfavorable entropic contribution, implying that enthalpic and entropic contributions equilibrated. The assembly of the negatively charged peptide (Asp6) with TA, on the other hand, was codriven by entropy and enthalpy and because |−TΔS| > |ΔH|, binding was dominated by entropy. In contrast, Pro6–TA and Tyr6–TA bindings showed an unfavorable enthalpy and a favorable entropy and were, therefore, entropy driven, which is consistent with hydrophobic association. Analysis of the ITC data indicates that although multiple interactions may exist between the TA and peptides, the enthalpy drive provided by electrostatic interactions is necessary to sustain the growth of LbL-assembled coatings of polyphenols and peptides. In addition, thermodynamic changes in the binding between the TA and positively charged His6 were investigated under acidic conditions (pH 3). The significant decrease in |ΔH| at pH 3 relative to pH 7 indicates that the electrostatic interaction between the TA and His6 is severely weakened under acidic conditions and leads to the decomposition of the assembled coating at pH 3. Therefore, positively charged peptides are a suitable choice for assembly with polyphenols to prepare multilayered coatings.
Fig. 3
Fitted curves for changes in molar enthalpy plotted as function of peptide-to-TA molar ratio obtained by titrating (a) Arg6, (b) His6, (c) Lys6, (d) Asp6, (e) Pro6, (f) Tyr6, and (g) His6 in TA at 25 °C and pH 3. (h) Summary of thermodynamic parameters (ΔH, ΔG, and − TΔS) determined from ITC analysis
Molecular dynamics simulation of TA and peptide assemblyTo further investigate the interaction between TA molecules and peptides, a series of all-atom molecular dynamics (MD) simulations were performed. As shown in Fig. 4a-c, initially (0 ns), the TA molecules and predicted peptides were placed randomly in a water box of 12.7 × 12.7 × 12.7 nm [3]. In about 90 ns from the all-atom MD simulation trajectory, most of the TA molecules aggregated and formed one major cluster. In 180 ns from the all-atom MD simulation trajectory, all the TA molecules aggregated and formed one cluster, the peptides are distributed around the TA molecules and interact with the TA cluster. This is consistent with the peptide-coated surface observed by LP-AFM and explains the reason why the TA with His6 assembly coating consists of particles. Subsequently, we performed a series of 500 ns all-atom MD simulations on the effects of an acidic solution environment on the interaction between the peptide composed of six histidine residues and TA molecules. The structure of the peptide composed of six histidine residues (His6 peptide) was predicted by AlphaFold2. In Fig. 4d, the mean square deviation (MSD) curve of His6 peptide and TA molecules in the acid (pH = 3) and neutral (pH = 7) solution environment in the 500 ns all-atom MD simulation showed that the TA molecules aggregated and formed one major particle in about 200 ns time scale. Only one His6 peptide was captured along the surface of aggregated TA molecules (Fig. 4e) and the MSD curve indicated that the His6 peptide tends to diffuse and depart from TA molecules in the acid (pH = 3) solution environment. On the other hand, a series of His6 peptides were captured along the surface of the aggregated TA molecules (Fig. 4f) and the MSD curve indicated that the His6 peptide tended to interact with TA molecules in the neutral (pH = 7) solution environment. This is consistent with the experimental results (Fig. 2b).
Fig. 4
(a-c) The snapshot (0 ns, 90 ns, 180 ns) from the all-atom molecule dynamics simulation trajectory showed the aggregation of TA molecules and peptides. The TA molecules were shown in licorice colored and the peptides were shown in new cartoon and silver. (d) The MSD time evolution of His6 peptide (dashed) and TA molecules (solid) in the acid (pH = 3, blue) and neutral (pH = 7, red) solution environment. (e) The interaction between His6 peptide (green) and TA molecules (magenta) in the acid (pH = 3) solution environment. (f) The interaction between His6 peptide (green) and TA molecules (magenta) in the neutral (pH = 7) solution environment. The atoms that may formed hydrogen bonds were shown in the yellow dashed line
Furthermore, the structures of the positively charged peptides (Lys6 and Arg6 peptide) and the negatively charged peptide (Asp6 peptide) were predicted by AlphaFold2, respectively, and a series of 500 ns all-atom MD simulations were carried out to study their interactions with TA molecules. In Figure S15a, the mean square deviation (MSD) evolution of Lys6, Asp6, and Arg6 peptide and TA molecules in a neutral (pH = 7) solution environment in the 500 ns all-atom MD simulation showed that the TA molecules aggregated and formed one major particle in about 300 ns time scale. Multiple positively charged Lys6 and Arg6 peptides can subsequently be captured along the surface of the aggregated TA molecule, whereas only one negatively charged Asp6 peptide can be captured (Figure S15b-d). Meanwhile the MSD curves indicated that positively charged peptides (Lys6 and Arg6 peptide) tended to be captured by the aggregated TA molecules while negatively charged peptides (Asp6 peptide) tended to diffuse and detach from the TA molecules. In addition, molecular dynamics simulations showed stronger interactions between TA and positively charged peptides relative to negatively charged peptides. This is consistent with the experimental results and illustrates the importance of electrostatic interactions for polyphenol-peptide assembly.
Owing to the complexity of protein structures and unclear interaction regions, interactions between proteins and polyphenols have always been difficult to elucidate. For example, previous studies have demonstrated electrostatic, hydrogen-bonding, and hydrophobic interactions between the lysozyme (Lyz) and TA [45]. Recently, we have found no covalent interactions during the assembly of TA and Lyz in the pH 6–8 range, and although the driving force for assembling both originates from favorable enthalpy changes due to noncovalent interactions, the exact interaction species from which the enthalpy changes originate is still unknown [26]. Interactions and thermodynamic processes between TA and peptides containing different specific sequences and exhibiting different properties enable a deeper understanding of the action mechanism between the lysozyme and TA. This study shows that although all types of peptides (charged, uncharged, and hydrophobic) interacted with TA, the favorable enthalpy change due to electrostatic interactions was the main driving force for sustaining the assembly of the peptide and TA coatings, and the favorable enthalpy change due to positively charged peptides (Arg6, His6, and Lys6) was much larger than that due to negatively charged peptides (Asp6). Similarly, although all regions of Lyz (charged, uncharged, and hydrophobic) can simultaneously exhibit multiple noncovalent interactions with TA, electrostatic interactions dominate between the positively charged regions (amino-acid sequences) and TA, and favorable enthalpy changes due to electrostatic attractions are the main reason for sustaining the growth of Lyz–TA films LbL, which clearly reveals the action mechanism during the LbL assembly of Lyz and TA and provides a new idea for understanding and regulating the interaction mechanism between proteins and polyphenols.
Cell proliferation and antioxidant effects of peptide coatingsPeptide coatings exhibit good hydrophilicity and, thus, provide suitable conditions for cell growth (Figure S16). Figure 5a and b show the graphical and quantitative analyses of the effect of the coating in which TA was assembled with the positively charged peptide (His6) on cell proliferation, respectively. Proliferation was observed for dental pulp stem cells cultured on (TA/His6)15-coated and control blank slides. Figure 5a shows the fluorescence microscopy images after 3 d of cell culture. The dental pulp stem cells grew well on both the control blank and (TA/His6)15-coated slides, and the cells cultured on the (TA/His6)15-coated slide were denser and more elongated than the cells cultured on the control slide, which indicates that the coating helped to differentiate the stem cells. On the (TA/His6)15 coating, cellular proliferation was quantified using the CCK-8 method to count the number of cells, as shown in Fig. 5b. Although the cell viability increased with prolonged incubation on both the control and (TA/His6)15-coated slides, the cells grew more rapidly on the coating and were significantly different from the cells that were cultured on the control slide, indicating that the (TA/His6)15 coating was beneficial for promoting cell proliferation. Although we hypothesized that the proliferative function of the coating might originate from the formation of a functional secondary structure after the peptide assembly, CD spectroscopy did not reveal any obvious peptide secondary structure before or after assembly (Figure S17). Therefore, the proliferative function was mainly derived from the activity and interaction between the TA and peptides.
Figure 5c shows the ABTS•+ removal kinetic curves obtained for various coatings in which TA was assembled with different positively charged peptides at pH 7. Relative to the blank control, TA/peptide-coated samples showed significant ABTS•+ radical scavenging. Although the final amounts of free-radical scavenging were similar for different coatings, the free radical–scavenging rates were substantially different, which may be caused by different interaction strengths between the TA and different positively charged peptides. Interactions between TA and Arg6, His6, and Lys6 gradually weakened, which accelerated the free-radical scavenging of the corresponding coatings. To further evaluate the intracellular antioxidant ability of the coatings, H2O2 was used to stimulate ROS formation because H2O2 can rapidly generate considerable ROS. As shown in Fig. 5d, compared with the nontreated cells, the H2O2-treated cells cultured on the glass slide (in the control group) were drastically reduced in both size and number, while the cells cultured on the (TA–His6)15-coated slide negligibly changed in both size and number. Statistical analysis of the number of cells and cellular area further confirmed these observations, as displayed in Fig. 5e and f, respectively. These results suggest that the (TA–His6)15 coating has strong antioxidant activity.
To visualize the level of intracellular ROS, cells on slides and coatings were stained (green) using an ROS detector and imaged before/after treatment with H2O2. As shown in Fig. 5g, cells cultured on blank slides were greener than those cultured on the (TA/His6)15 coating both before and after H2O2 treatment. Cells cultured on blank slides became brighter after H2O2 treatment, while the color of cells cultured on the (TA/His6)15 coating negligibly changed. The mean fluorescence intensity of each cell was analyzed using ImageJ software, as shown in Fig. 5h. The mean fluorescence intensity of each cell significantly increased for cells cultured on blank slides and negligibly changed for cells cultured on the (TA/His6)15 coating. To detect further differences in intracellular ROS levels, flow cytometry, which is more sensitive to fluorescence intensity, was used, and the percentage of positive cells was calculated and analyzed using the accessory software, as shown in Fig. 5i. After H2O2 treatment, the percentage of positive cells increased in all the samples. However, cells cultured on blank slides showed a much higher ploidy than those cultured on the (TA/His6)15 coating. These results show that the (TA–His6)15 coating has good ROS resistance. Notably, (TA/Lys6)15 coating prepared by the same method exhibited better cell proliferation (Figure S18) and cellular antioxidant effects (Figure S19), which provided a superior carrier for subsequent functional customization of the peptide coating, further validating the universality of the polyphenolic peptide functional coating preparation strategy.
Fig. 5
(a) Fluorescence microscopy images of dental pulp stem cells seeded on glass and (TA/His6)15-coated slides for 3 d. (b) Relative activities of dental pulp stem cells seeded on glass and (TA/His6)15-coated slides for 1, 2, and 3 d. (c) Time evolution for quantity of ABTS•+ reacted with (TA/hexapeptide)15 coating. (d) Fluorescence microscopy images of dental pulp stem cells seeded on (TA/His6)15-coated slides before and after H2O2 treatment. (e) Area per cell and (f) number of cells per mm [2] on (TA/His6)15-coated slides before and after H2O2 treatment. (g) Fluorescence microscopy images of ROS detector inside dental pulp stem cells seeded on (TA/His6)15-coated slides before and after H2O2 treatment. (h) Mean fluorescence intensity per cell for cells seeded on (TA/His6)15-coated slide before and after H2O2 treatment. (i) Positive rate of intracellular ROS levels generated based on cytometry data
Multilayered capsules assembled from TA and peptidesAs shown in Fig. 6a, PS was used as a template, and multilayered particles were prepared using TA and Lys6 building blocks and LbL assembly. Then, the template was removed using THF to obtain multilayered capsules. The surface potential and particle size of the multilayered particles and capsules were monitored during LbL assembly (Fig.
留言 (0)