Materials
Ce6 was purchased from Glpbio Technology Inc. (USA). TPP, 3-Bromopropylamine hydrobromide, EDC, NHS, Cupric sulfate, DMSO-d6, MTT, and other required chemical reagents were obtained from Aladdin Biochemical Technology Co., Ltd. (Shanghai, China). TP5 was purchased from Meilunbio Technology Co., Ltd. (Guangzhou, China). Annexin V-FITC Apoptosis Detection Kit and d-Luciferin, Potassium Salt were purchased from Yeasen Biotechnology Co., Ltd. (Shanghai, China). LDH Cytotoxicity Assay Kit, Enhanced mitochondrial membrane potential assay kit with JC-1, GSH, and GSSG Assay Kit, ROS Assay Kit, Crystal Violet Staining Solution, and ATP Assay Kit all were acquired from Beyotime Biotech. Inc. (China). Cell-Light EdU Apollo488 In Vitro Kit was purchased from Ruibo Biotechnology Co., Ltd. (China). MitoTracker Green FM and Lipofectamine 3000 kits were obtained from Invitrogen (USA). IL-2, IL-6, IL-10, TNF-α, and IFN-γ ELISA kits were purchased from ABclonal Technology Co., Ltd. (Wuhan, China). Granzyme B Elisa Kit was acquired from Solarbio Science & Technology Co., Ltd. (Beijing, China). Antibodies for flow cytometry were purchased from Biolegend (Beijing, China). The experiment utilized Abcam’s provided anti-ATP7A (ab308524) and anti-AMPK (ab92701), Zenbio’s provided anti-GAPDH (380,626) and anti-CRT1 (510,109), Abclonal’s provided anti-Phospho-AMPKα (AP1002), anti-PD-L1 (A1645), and anti-cGAS (A8335), Proteintech’s provided anti-DLAT (68,303-1-Ig), and anti-FDX1 (12,592-1-AP), CST’s provided anti-STING (13647S), anti-IRF-3 (4302S), anti-Phospho-IRF-3 (29047S), and anti-PGP (13978S). Besides, anti-lipoic acid (sc-101354) was provided by Santa Cruz Biotechnology, Inc., anti-Ki67 (bs-23103R) was purchased from Beijing Biosynthesis Biotechnology Co., Ltd., and anti-ATP7B (A00686-2) was obtained from Boster Biological Technology Co., Ltd. (China).
Cell lines and animals
U87 and bEnd.3 cells were sourced from the American Type Culture Collection, while GL261 and GL261-luc cells were obtained from Sunncell Biotechnology Co., Ltd. (China). The cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM; Hyclone, USA), supplemented with 10% fetal bovine serum (FBS, Gibco, USA), and 1% penicillin–streptomycin solution (Beyotime, China) at 37 °C in a humidified atmosphere containing 5% CO2.
Owing to male mice being more suitable to survive during the establishment of orthotopic GBM models, male C57BL/6 mice (5–6 weeks old, 18–22 g) were purchased from Gempharmatech Co., Ltd. (Chengdu, China). The mice were housed in an SPF-grade animal facility with a 12-h light–dark cycle at 25 °C and controlled humidity.
Synthesis of TPP-NH2 and TCe6
To synthesize TPP-NH2, 5 mM TPP and 5 mM 3-Bromopropylamine hydrobromide were dissolved in a total of 20 mL anhydrous acetonitrile. The mixture was stirred for 15 h at 85 °C using an oil bath equipped with a reflux apparatus. After the reaction was complete, the solution was cooled to ambient temperature, and the crude product TPP-NH2 was collected via a rotary evaporator. Subsequently, the crude product underwent recrystallization using a composite solvent system consisting of 3.4 mL anhydrous acetonitrile, 10 mL n-Hexane, 34 mL isopropanol, and 17.5 mL diethyl ether. Finally, the purified TPP-NH2 was obtained by suction filtration.
For the synthesis of TCe6, TPP-NH2 was conjugated to Ce6 via an acylation reaction of aminocarboxylic acids. Ce6 (50 mg, 27.9 mM) was dissolved in 3 mL methyl alcohol. EDC (32 mg, 55.6 mM) and NHS (19.3 mg, 55.9 mM) were added to this solution under magnetic stirring with protection from nitrogen and an ice bath over 30 min. After careful removal of the ice, purified TPP-NH2 (161 mg, 167.54 mM) was added under magnetic stirring with nitrogen protection for 24 h. The crude product TCe6 was then collected via a rotary evaporator and purified using silica gel column chromatography with a gradient elution ratio of 100: 1 to 10: 1 (dichloromethane: methanol). The final products TPP-NH2 and TCe6 were analyzed by 1H-NMR and mass spectrometry.
Preparation and characterization of TCe6@Cu/TP5
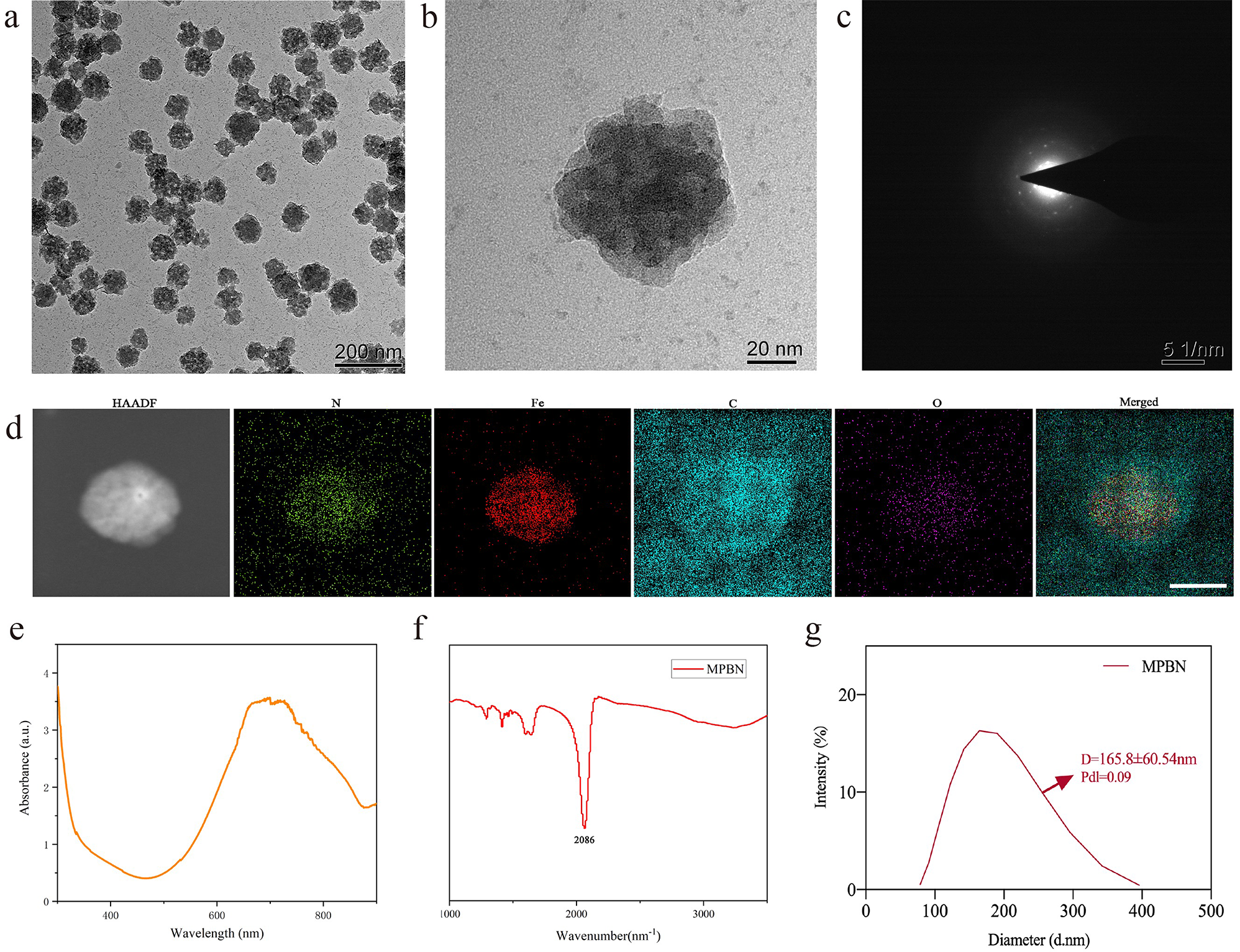
To prepare TCe6@Cu, 1 mg TCe6 and 2 mM Cu (CuSO4), dissolved in methanol, were stirred at room temperature for 30 min. To prepare TCe6@Cu/TP5, 1 mg TCe6, 2 mM Cu, and 2 mg TP5 were stirred in methanol solution for 30 min. Subsequently, TCe6@Cu/TP5 was collected in ultrapure water via the film dispersion method after the elimination of methanol by rotary steaming. The precipitate was recovered by centrifugation (10,000 g, 4 ℃, 10 min) using a Dragon Lab D3024R centrifuge, and the collected precipitate was washed twice with 5% methanol of ultrapure water. The final collected precipitate was diluted in 1 mL PBS solution, followed by sonication in an ice bath for distribution. TCe6@Cu was prepared using the same procedure as described above.
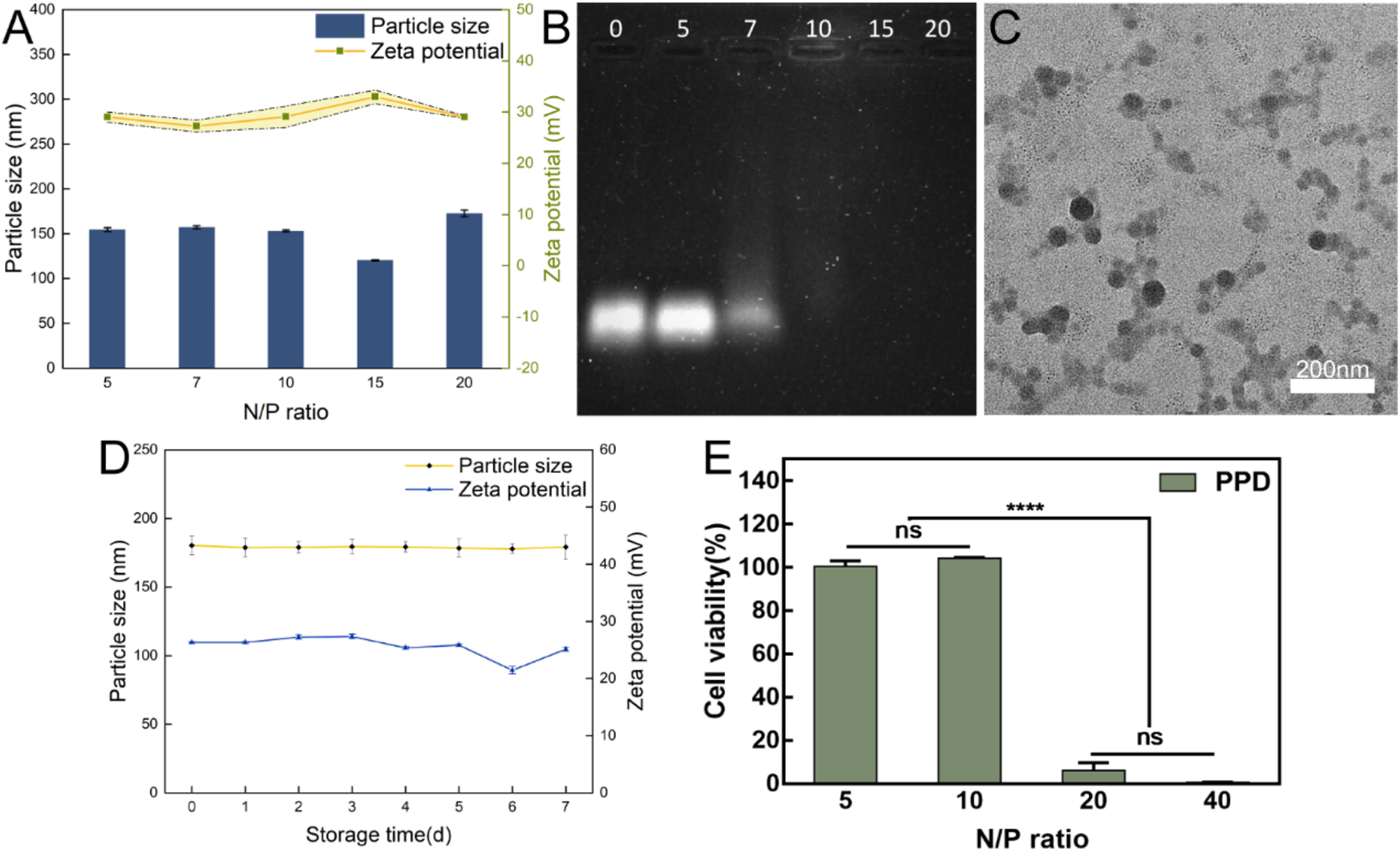
The particle size and zeta potential of TCe6@Cu and TCe6@Cu/TP5 were measured using a Malvern ZetaSizer Nano series (Westborough, MA). Transmission electron microscopy (TEM) images were captured with a TEM (Hitachi High-Tech, HT7800). The presence of TP5 in TCe6@Cu/TP5 NPs was confirmed through Coomassie brilliant blue staining using a 13% SDS-PAGE gel. UV–vis absorption spectra of Ce6, TCe6, TCe6@Cu, and TCe6@Cu/TP5 at room temperature were recorded using ultraviolet–visible spectroscopy (Shimadzu, UV-2600i). Fluorescence spectra were obtained using Spark (Tecan) for Ce6 and TCe6 diluted in methanol, while TCe6@Cu and TCe6@Cu/TP5 were dissolved in ultrapure water. The concentrations of TCe6 and TP5 were determined by UV − vis absorption spectra, and the concentration of copper was determined by ICP-OES (Agilent, 5100 SVDV). Additionally, the surface analysis of TCe6@Cu/TP5 was conducted using X-ray photoelectron spectroscopy (Thermo Scientific, K-Alpha). The serum stability of TCe6@Cu/TP5 was measured by Malvern ZetaSizer Nano series, with the sample diluted in 10% DMEM containing 10% FBS.
Singlet oxygen measurement
To assess the production of singlet oxygen under NIR irradiation, a singlet oxygen probe DPBF was employed. Specifically, 30 μL of DPBF (1 mg·mL−1, dissolved in DMF) was added to 2 mL of water, as well as to solutions of Ce6, TCe6, TCe6@Cu, and TCe6@Cu/TP5 TCe6@Cu/TP5 NPs. Subsequently, free DPBF, Ce6, TCe6, TCe6@Cu, and TCe6@Cu/TP5 TCe6@Cu/TP5 NPs were subjected to 660 nm NIR irradiation (1.0 W cm−2) for varying durations (0, 10, 20, 30, 40, 65 s). The UV–vis spectrometer was utilized to scan wavelengths ranging from 350 to 550 nm.
Copper ions responsive release evaluation
A TEM image of TCe6@Cu/TP5 NPs containing 10 mM GSH was captured using RuliTEM. Subsequently, an MB (50 µM) solution containing TCe6@Cu/TP5 NPs was mixed with 10 mM GSH and 10 mM H2O2. The production of·OH was quantified based on the degradation of MB, and the mixture containing GSH for 2 h was analyzed using UV–vis absorption spectroscopy.
Furthermore, colorless DTNB transforms into yellow 5-mercapto-2-nitrobenzoic acid in the presence of a sulfhydryl compound (-SH), exhibiting maximum absorption at 412 nm. To assess this, 10 μL DTNB solution (200 μM) was added to 200 μL GSH (1 mM), TCe6@Cu/TP5 NPs, and the mixture of GSH and TCe6@Cu/TP5 NPs. The absorbance at 412 nm (A412 nm) was measured after 30 min.
Drug responsive release evaluation
TCe6@Cu/TP5 NPs enclosed in dialysis bags (3,500 kDa) were immersed in PBS solution at different pH values (pH 7.4 and 5.0) containing varied concentrations of GSH (0 and 10 mM) along with 5% Tween-80. The setup was subjected to shaking at 150 rpm on a horizontal laboratory shaker at 37 ℃. At specified intervals (0.5, 1, 2, 4, 6, 8, 12, 24, 36, 48, 60, and 72 h), 1 mL of the supernatant was withdrawn. A UV- vis spectrometer determined the quantity of released TCe6 from the samples.
Detection of cellular uptake
GL261 and U87 cells were seeded at a density of 5 × 105 cells/well in a six-well culture plate and incubated for 24 h. Subsequently, the medium was exchanged with 1 mL of DMEM containing TCe6@Cu/TP5 NPs (5 μM of TCe6), and the cells were further incubated for 0, 1, 2, 4, 6, and 8 h at 37 ℃. Following incubation, cells were rinsed twice with PBS, fixed with 4% paraformaldehyde, and assessed for cellular uptake using fluorescence imaging analysis (Olympus, Tokyo, Japan) and flow cytometry analysis (BD FACS Celesta).
Cytotoxicity assay of MTT and LDH
For the viability assay, GL261 and U87 cells (5 × 103 cells/well) were plated in 96-well plates and cultured for 24 h. Subsequently, the cells were treated with PBS, TP5, TCe6, TCe6@Cu, and TCe6@Cu/TP5 NPs for 4 h. Following drug withdrawal, with or without laser irradiation (660 nm laser at 1.0 W cm−2) for 1 min per cell, the cells were further incubated for 24 h. MTT solution (5 mg mL−1) was then added and incubated for 4 h. After removing the culture medium, 150 μL DMSO was added to dissolve the produced formazan crystals, and the absorbance at 562 nm was measured.
For the LDH release assay, GL261 and U87 cells were seeded into 96-well plates for 24 h. In the case of GL261 cells, various treatments, including PBS, TCe6 (2 μM), TCe6@Cu (containing 2 μM TCe6), TCe6@Cu/TP5 NPs (also comprising 2 μM TCe6), and TP5 (at an equal concentration to TP5 within TCe6@Cu/TP5 NPs), were applied for 4 h post-dosing. After discontinuation of the treatments, laser exposure was performed, followed by an additional 24-h incubation. The treatment of U87 cells mirrored that of GL261 cells but with halved concentrations of all agents. Each well was then collected, mixed with LDH reagent, and left in darkness for 30 min. The light absorption value of LDH was measured using an enzyme-label instrument.
Cell proliferation assay of EdU and colony formation
In the EdU assay, cells were seeded into 96-well plates and treated as indicated. Active cell proliferation was assessed using the Cell-Light EdU Apollo488 In Vitro Kit (C10310-3).
For the colony formation assay, a density of 5 × 104 cells per well was seeded in 12-well plates for approximately 48 h and treated with the indicated agents as before. After treatment, the cells were cultured for an additional 72 h. Images were captured after fixation with 4% paraformaldehyde for 2 h, staining with 0.2% crystal violet for 1 h, and washing three times with PBS.
In vitro apoptosis assay
To evaluate the apoptosis of GL261 and U87 cells, GL261 and U87 cells (5 × 105 cells per well) were seeded in six-well plates for 24 h separately. The indicated agents were added as in the LDH assay. Stain the cells with Annexin V-FITC and PI or JC-1 solution according to the instructions, and detect cells by flow cytometry.
In vitro siRNA transfection assay
ATP7A, CTR1, PGP, and silencer negative control siRNAs were obtained from Beijing Tsingke Biotech Co., Ltd. The sequences were designed using the RNAi designer tool from Thermo Scientific, and they are as follows: ATP7A siRNA, 5’-CACUGUUGAGGGAAUGACAUGUAUU-3’; CTR1 siRNA, 5’-ACGACAACAUU -ACCAUGCCACCUCA-3’; PGP siRNA, 5’-CGACUGGGCUUCAUCACCAACAA -CA-3’. The bEnd.3 cell lines were cultured in DMEM supplemented with 10% FBS until cells reached approximately 50% to 70% confluence in each six-plate well. For siRNA transfection, bEnd.3 cells were transfected with 50 nM siRNAs using lipofectamine 3000 (L3000015) for 6 h. After that, transfected cells were placed into a growth medium and incubated for 48 h at 37 ℃ before experiments. Western blotting was used to determine the efficiency of siRNA transfection.
In vitro BBB transcytosis assay
Cell lines bEnd.3 were used to construct an in vitro BBB model, following a previously reported protocol [24, 25]. These cells were uniformly plated at a density of 6 × 104 cells·cm−2 at 37 ℃ on 0.4 µm pore transwell polycarbonate membranes (Corning, 3342), achieving confluence within 48 h. Subsequently, the lower chambers contained blank medium, GL261 cells, or U87 cells, while respective solutions of Ce6, TCe6, and TCe6@Cu/TP5 NPs (4 µM) were introduced apically. Additionally, TCe6@Cu/TP5 NPs were added to the upper chamber with the transfected bEnd.3 cells. BBB transcytosis was evaluated through fluorescence imaging analysis, fluorescence spectroscopy analysis, and/or flow cytometry analysis.
Furthermore, bEnd.3 cells were seeded at a density of 5 × 103 cells per 96-well plate for 24 h. TCe6@Cu/TP5 NPs (4 µM) were added and incubated for 0, 2, 4, 8, 10, 12, and 24 h at 37 ℃ in a humidified atmosphere containing 5% CO2. Assessments of TCe6@Cu/TP5 NPs crossing the BBB were conducted using fluorescence images and fluorescence spectroscopic analysis.
In vitro mitochondrial co-localization assay
GL261 and U87 cells, freshly plated in confocal dishes, were treated for 4 h following a 24-h incubation. After two rounds of PBS treatment, mito-tracker solution (1:10,000) was added and incubated at 37 ℃ for 30 min. Subsequently, the stained cells were observed using Nikon's AXR laser confocal microscopy to assess specific targeting activity towards mitochondria.
In vitro GSH/GSSG ratio measurement
GL261 and U87 cells, evenly dispersed in six-well plates, underwent post-treatment collection. The measurement of GSH vs GSSG level utilized the GSH/GSSG Assay Kit (S0053), adhering strictly to the manufacturer's protocol.
Intracellular ATP consumption assay
GL261 and U87 cells were incubated for 24 h in the six plates and treated as before. The cultured medium was discarded. Then, according to the instrument of the ATP assay kit, intracellular ATP levels were determined using a luminometer in a black 96-well plate.
In vitro ROS generation
GL261 and U87 cells were seeded into 6-well plates and incubated for 24 h. After being treated with the indicated agents for 4 h and laser exposed for 2 min. Following two PBS washes, cells were suspended in a serum-free medium while 10 µM DCFH-DA was added for 15 min at 37 ℃. Images were taken under fixed conditions using 4% paraformaldehyde.
Western blotting assay
Protein extraction was carried out using RIPA buffer with protease inhibitor cocktails (A, B, C) (Invitrogen, USA). The protein concentration was quantified using the BCA assay kit (Tiangen, China). Proteins were separated by SDS-PAGE on 8%, 11%, or 15% gels and transferred to PVDF membranes (Millipore, USA). Primary antibodies (1:1000 for cGAS, STING, IRF-3, p-IRF-3, AMPK, p-AMPK, PD-L1, ATP7A, PGP, FDX1, and DLAT; 1:500 for CRT1; 1:3000 for GAPDH; 1:100 for lipoic acid) were incubated with the treated PVDF membranes overnight at 4 ℃. After three washes with TBST, membranes were exposed to secondary antibodies for 90 min. Detection was performed using an ECL Kit (Millipore, USA), with GAPDH serving as the internal standard.
In vitro co-cultured assay
GL261 and U87 cells were exposed to the specified concentrations of PBS, TP5, TCe6, TCe6@Cu, and TCe6@Cu/TP5 NPs solutions in six-well plates. After laser irradiation for 2 min and incubation for 24 h, primary culture cells from the spleen of C57BL/6 mice were evenly added to these GBM cells for co-culture for an additional 48 h. Subsequently, red blood cells from the upper layer of the spleen cells were eliminated using red blood cell lysis buffer. Following this, the cells were washed twice and stained with fluorescently labeled antibodies targeting positive CD4+ T cells, CD8+ T cells, and DC cells. Finally, the stained cells were detected via flow cytometry and analyzed using FlowJo software.
In vivo brain biodistribution assay
1 × 106 GL261-luc cells were intracranially inoculated into the brains of C57BL/6 mice (6-week-old) to establish an orthotopic brain tumor model. Ten days ago, the brain distributions of TCe6, TCe6@Cu, and TCe6@Cu/TP5 NPs in vivo were evaluated by fluorescence imaging taken at 2, 4, 6, 8, and 24 h using An IVIS Lumina XRIII system. After the experiment, the main organ tissues (brain, heart, liver, spleen, lung, and kidney) of these mice were imaged in vitro.
In vivo anti-orthotopic brain tumor study
Male C57BL/6 tumor-bearing mice (n = 5) were assigned into five groups. Groups that received i.v. injection of PBS, TP5 (2.7 mg·kg−1), TCe6 (2 mg·kg−1), TCe6@Cu (including 2 mg·kg−1 TCe6), or TCe6@Cu/TP5 NPs (including 2 mg·kg−1 TCe6) were irradiated with NIR light for 2 min post-tail vein insertion for 6 h. The tumor progression was tracked through tumor volume measurements via bioluminescent imaging at designated times. Before imaging, d-fluorescein (150 mg·kg−1) was intraperitoneally administered to induce imaging signals. The resulting signal was quantified as average photons per second per square centimeter per steradian. Then, mice were sacrificed and the brains within tumors, organs, and blood were harvested.
Cerebral and organ tissues were fixed in 4% paraformaldehyde for H&E staining and IHC immunohistochemistry analysis. Tumor sections were assayed with rabbit anti-Ki67, anti-PD-L1, anti-DLAT, anti-FDX1, anti-cGAS, and anti-STING antibodies at 4 ℃ overnight, followed by HRP-conjugated secondary antibody treatment for 40 min. The copper level of blood was determined using a Copper Colorimetric Assay Kit (Elabscience), while serum markers of alanine aminotransferase (ALT), aspartate aminotransferase (AST), urea, as well as creatinine (CRE) were evaluated via the cobas c311 analyzer (Roche). Besides, spleen tissues were used to detect CD4+ T cells, CD8+ T cells, Treg cells, and DC cells by flow cytometry and analyzed by FlowJo software.
In vivo anti-bilateral tumor study
Injected 2 × 106 GL261 cells into the flanks of C57BL/6 mice respectively. Once the tumor volume reached 100 mm3, the mice were randomly divided into five groups, providing various agents intravenously into the left flanks (n = 4 each) for anti-tumor assessment. Subsequently, the left side of the mice was illuminated with NIR light for 2 min after 2 h injection. The administrations were performed at specific times. Mouse weights were documented, and tumor volumes were calculated using the formula (Length × Width2/2). Tumor growth curves and average volumes for each group and individual mice were built. The mice were humanely euthanized and tissues, including tumors, organs, and blood, were collected.
Fixed right tumors in 4% paraformaldehyde for H&E staining. To evaluate biosafety, automatic blood biochemistry instruments were used to test ALT, AST, UREA, and CRE. To explore the immune mechanism, a flow cytometry assay was performed to detect CD4+ T cells, CD8+ T cells, Treg cells, and memory T cells in the spleen and DC cells in the lymph nodes. ELISA assay was performed to determine the level of IL-2, IL-6, IL-10, Granzyme B, TNF-α, and IFN-γ following the manufacturer’s protocols.
Statistical analysis
Statistical analysis using GraphPad Prism 8.0 revealed results expressed as mean ± standard deviation (SD). One-way ANOVA and Linear Regression were utilized for statistical comparisons with significance (* for p < 0.05, ** for p < 0.01, *** for p < 0.001, and **** for p < 0.0001).


留言 (0)