
記住我
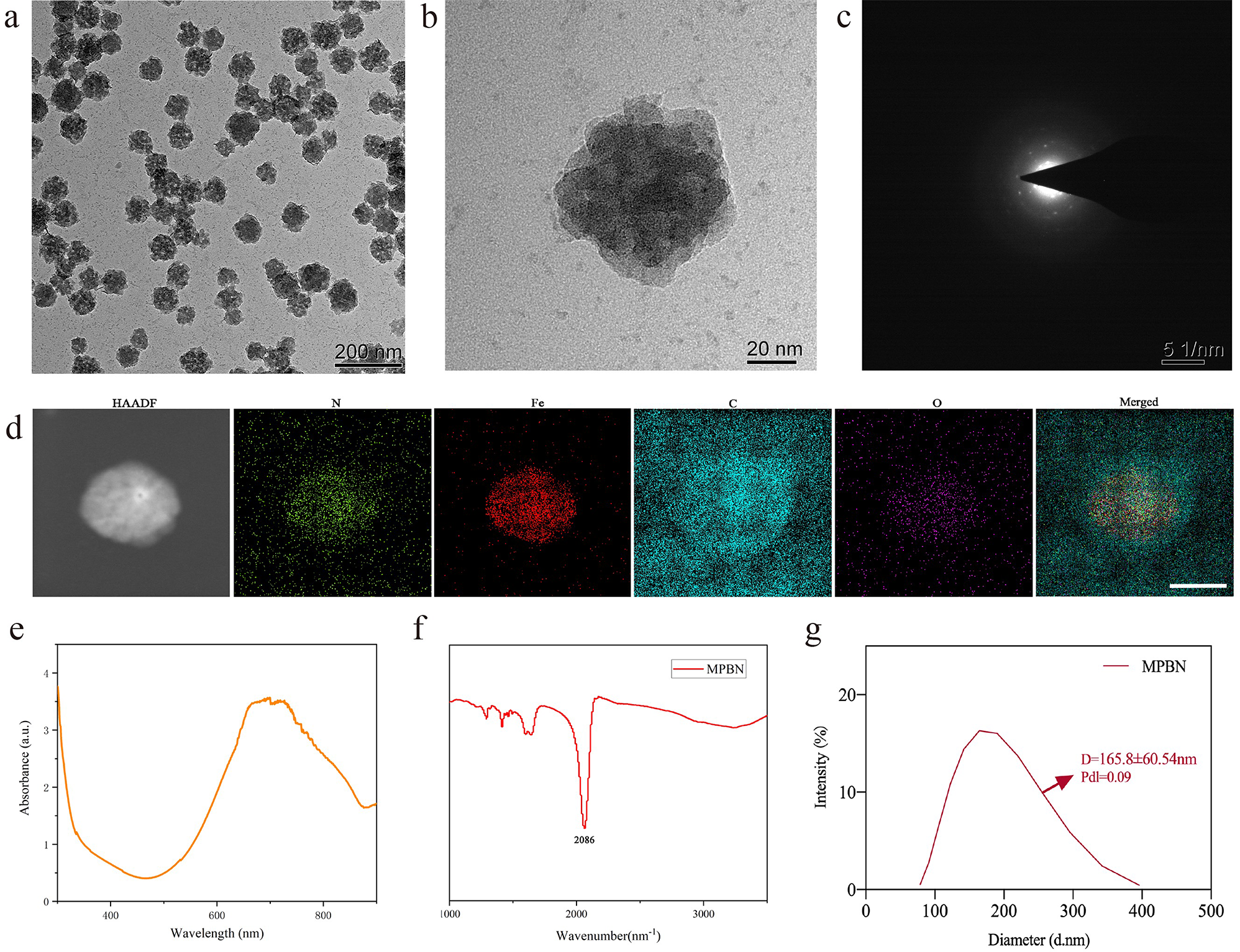
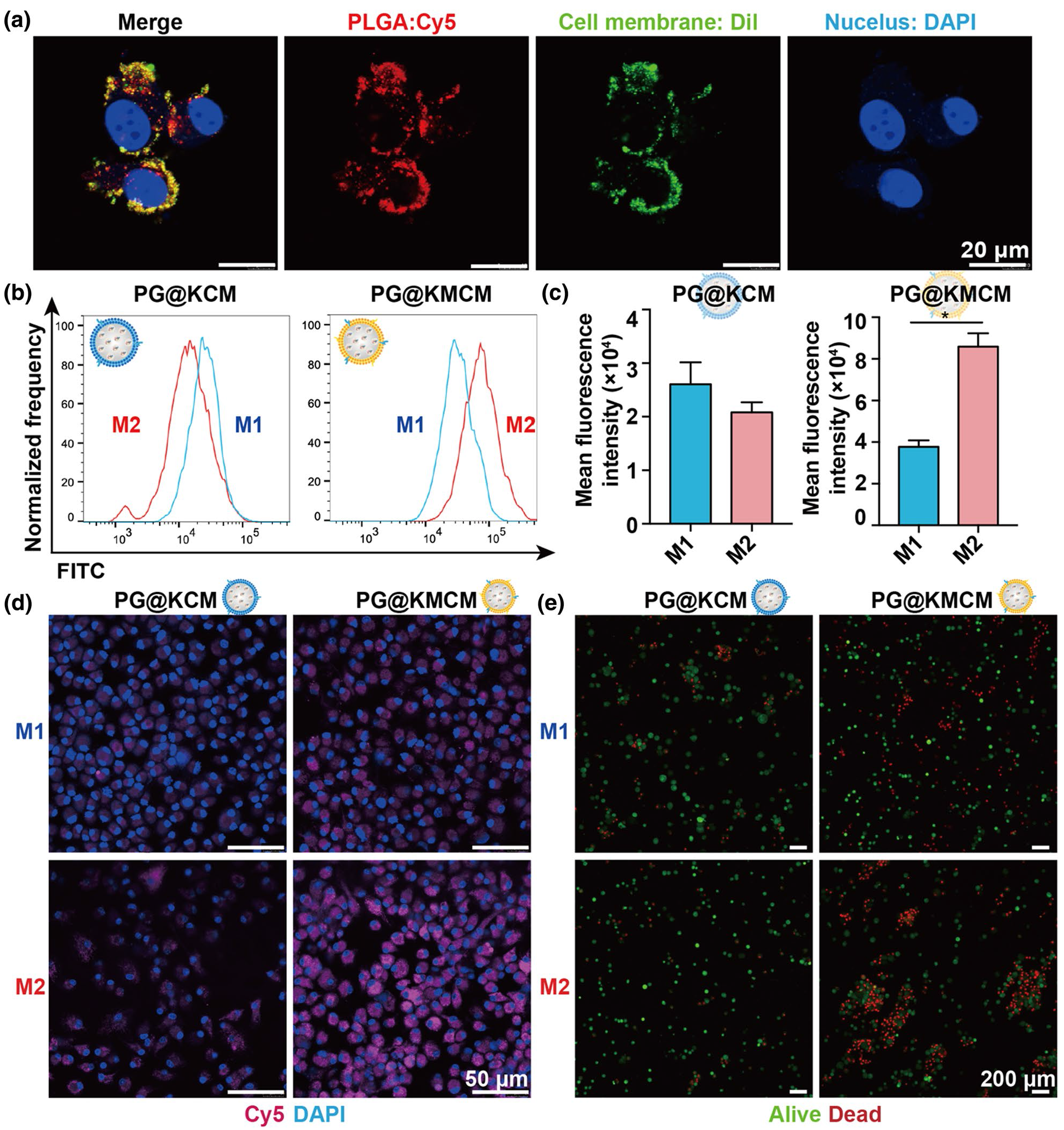

Mouse RAW264.7 cells (SNL-112) were purchased from the Shanghai Institute of Cell Biology, Chinese Academy of Sciences. The cells were cultured in Dulbecco’s Modified Eagle Medium (D5030, Sigma-Aldrich, USA) supplemented with 10% fetal bovine serum (10100147, Invitrogen, USA) and 1% penicillin/streptomycin (10378016, Invitrogen, USA) under conditions of 37 °C and 5% CO2. Upon reaching confluence, adherent cells were detached using a cell scraper. Approximately 1 × 107 cells were collected and centrifuged, and the pellet was resuspended in PBS buffer to obtain a single-cell suspension for subsequent preparation of NVs.
Macrophages were exposed to a hypotonic solution to disrupt the cell membrane, and morphological changes before and after rupture were observed under bright-field microscopy. Subsequently, a Dounce homogenizer was used to disrupt cell structures fully. The membranes were collected by density gradient centrifugation and stored at – 80 °C for further experiments [25].
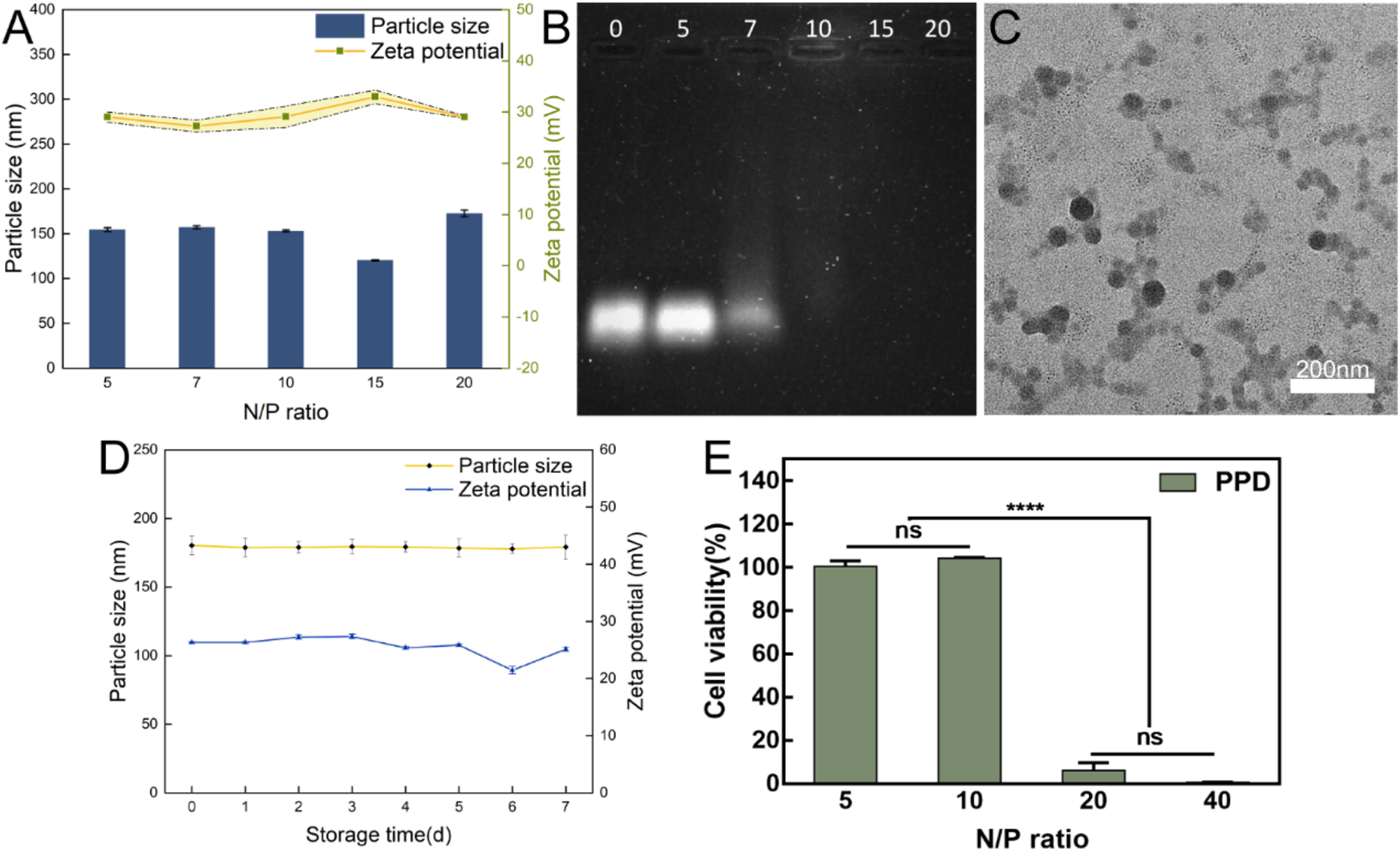
Isolation of NVs using continuous extrusion methodNVs were isolated from macrophages (RAW264.7) using a continuous extrusion method. In brief, after 3 days of cell culture, the cell suspension was subjected to extrusion using a small extruder machine (Avanti Polar Lipids, USA) equipped with a 100 nm pore size polycarbonate (PC) membrane filter (Whatman, UK). The suspension was extruded 5 times successively through PC membranes with pore sizes of 10 μm, 5 μm, and 1 μm. Subsequently, centrifugation was carried out at 2000 g and 20,000 g for 15 min each. Following a wash in PBS solution (centrifuged at 200,000 g for 2 h), the NVs were resuspended in a 5% trehalose PBS buffer solution (C1049, Beijing Baoosen Biological Technology Co., Ltd).
Preparation of macrophage membrane NVs-CURCUR loading into NVs was achieved using sonication as follows: Initially, a stock solution of 30 mg/mL CUR (C7727, Sigma-Aldrich, USA) was prepared in dimethyl sulfoxide (DMSO). Various concentrations of CUR (0.14, 0.42, 0.56, and 0.69 mg/mL) were added to 0.28 μg/μL NVs, with a PBS to DMSO mixture ratio of 2:1 (v/v). Three methods were evaluated for encapsulating CUR into NVs: freeze–thaw cycles, sonication, and room-temperature incubation. In the freeze–thaw method, the CUR solution was mixed with NVs, incubated for 30 min, rapidly frozen at – 80 °C for 8 min, and then thawed at room temperature; this freeze–thaw cycle was repeated three times. For sonication, the drug-NV mixture was sonicated for 15 min (at a frequency of 2 kHz, power of 70%) (KQ-600 KDE, Kunshan Ultrasonic Instruments Co., China), cooled at − 20 °C for 5 min, followed by another round of sonication. In the case of room temperature incubation, the drug-NV mixture was incubated at room temperature for 2 h. The purification of NVs-CUR was carried out using a two-step gradient centrifugation process. Initially, centrifugation at 8000 g for 15 min at 4 °C was performed to separate unloaded CUR, followed by centrifugation at 20,000 g for 15 min to obtain a brownish precipitate, identified as NVs-CUR, which was then resuspended in PBS.
Characterization and stability analysis of NVs-CURTransmission Electron Microscopy (TEM): 20 µl of NVs were dropped onto a copper grid and allowed to stand for 3 min. The liquid was then gently absorbed from the side using filter paper, followed by the addition of 30 μL of phosphotungstic acid solution (pH 6.8) (79690, Merck, USA). After incubating at room temperature for 5 min, the sample was air-dried under incandescent light. Subsequently, observation was conducted using a transmission electron microscope (JEM-1011, JEOL, Tokyo, Japan) under an accelerating voltage of 80 kV, and images were captured utilizing the side-mounted Camera-Megaview III (Soft Imaging System, Münster, Germany).
Nanoparticle Tracking Analysis (NTA): NVs samples were resuspended in PBS and diluted 500 times with Milli-Q water. The diluted NVs were then injected into the sample chamber of the NanoSight LM10 (Malvern, UK) instrument using a sterile syringe to ensure the absence of air bubbles and to fill the chamber completely. The videos were analyzed using NanoSight version 2.3 (Malvern, UK) software with a gain of 6.0 and a threshold of 11. The software tracked the motion of particles, outputting concentration and size distribution plots for the diluted sample, from which the original concentration of NVs was calculated based on the dilution factor.
Identification of NVs surface markers with Western blot: NVs were resuspended in RIPA lysis buffer (R0010, Solarbio, Beijing, China) and subjected to Western blot analysis to detect surface markers including CD9, CD81, CD63, and Alix, as well as the endoplasmic reticulum marker Calnexin. Antibody information for each marker can be found in the subsequent Western blot section. Each experiment was repeated three times [26].
Determination of drug loading efficiencyTo quantitatively determine the amount of CUR incorporated, purified NVs-CUR were collected, and the absorbance of CUR at 436 nm was measured using a UV–Vis spectrophotometer (U-3900, Hitachi, Japan). Subsequently, the respective CUR concentration in each NVs-CUR sample was calculated based on CUR’s calibration curve. Next, the drug encapsulation efficiency (EE) and loading capacity (LC) of NVs were calculated according to the following equations: EE = CNVs-CUR/Cinitial; LC = CNVs-CUR/(Cinitial + NNVs). Here, CNVs-CUR represents the amount of CUR in NVs-CUR, Cinitial is the initial amount of CUR added to the formulation, and NNVs is the amount of NVs.
Drug release kinetics determinationA specific amount of FITC NVs-CUR was added to a release medium of physiological saline, and the temperature was set to 37 °C to mimic human body conditions. At predetermined time points (0, 1, 2, 4, 8, 12, 24, 48 h), samples of a certain volume were withdrawn from the release medium, and the concentration of CUR in the release medium was measured using a UV–visible spectrophotometer (ND-1000, Nanodrop, Thermo Fisher, USA). The fluorescence intensity of FITC is correlated with the concentration of CUR, allowing the determination of the release rate through colorimetric analysis. By comparing the released concentrations at different time points with the initial loading amount, the proportion of CUR released within 48 h was calculated [27].
Cellular uptake of drug-loaded NVsIn order to investigate the internalization and intracellular distribution of NVs-CUR, Dil-labeled NVs-CUR were utilized to examine the uptake by PC12 cells (SNL-124, obtained from the Chinese Academy of Sciences Cell Bank). 1,10-Dioctadecyl-3,3,30,30-tetramethylindocarbocyanine perchlorate (Dil) (465498, Sigma-Aldrich, USA) was employed as a fluorescent dye to label NVs-CUR. Dil red fluorescent cell membrane probe (10 mM) was added to the suspension of NVs-CUR and incubated for 20 min to prepare Dil-labeled NVs-CUR. PC12 cells were cultured overnight in a confocal laser dish. When the cell density reached 70–80%, Dil-labeled NVs-CUR was introduced into the culture medium and co-incubated with PC12 cells. DAPI was used to stain the cell nuclei. At 1, 3, and 6 h post-incubation, the distribution of Dil-labeled NVs-CUR in PC12 cells was observed using a confocal laser scanning microscope (Synergy-2, BioTek Instruments, Winooski, Vermont, USA).
Cell viability and toxicity assays of nucleic vesicles (NVs)PC12 cells were cultured in DMEM supplemented with heat-inactivated 10% FBS and 1% penicillin/streptomycin and then incubated at 37 °C in a humidified atmosphere containing 5% CO2. Initially, the cytotoxicity of blank NVs at concentrations of 5–25 mg/mL on PC12 cells was evaluated using the MTT assay. PC12 cells were seeded in a 96-well plate at a density of 104 cells per well. Upon cell adhesion, NVs-CUR were diluted in the culture medium to concentrations of 5, 10, 15, 20, and 25 mg/mL. The culture medium served as the blank control. After 48 h of cell treatment, 20 µl MTT solution (5 mg/mL, 0.5% MTT) from Sigma-Aldrich was added to each well and incubated for 4 h. Subsequently, the liquid in the wells was aspirated and replaced with 150 µl DMSO. After shaking at low speed for 10 min, the optical density was measured at a wavelength of 490 nm using a microplate reader to calculate cell survival rates.
To assess the protective effect of NVs-CUR on PC12 cells, the influence of H2O2 (HX0640, Supelco, USA) on PC12 cell viability was determined using the MTT assay. Four hours after the H2O2 addition, PC12 cells were treated with PBS, CUR, and NVs-CUR. Subsequently, PC12 cell viability and proliferation were assessed using the MTT assay, with CUR and NVs-CUR both applied at a concentration of 100 ng/mL. Axonal growth of each well was observed and captured under a Nikon ECLIPSE80i (Nikon, Japan) microscope after 5 days.
Construction of lentivirus vectors for silence and overexpressionThe potential short hairpin RNA (shRNA) target sequences for mouse cDNA were analyzed based on GenBank. Three sequences targeting PCBP2 and SLC7A11 were designed, along with a negative control lacking interfering sequences (sh-NC), with primer sequences listed in Table S1. The oligonucleotides were synthesized by Genewiz and used to construct a lentivirus packaging system via lentivirus interference vector LV-1 (pGLVU6/GFP) (C06001, Genewiz, China).
Human embryonic kidney 293t cells (HEK293T) cell line was obtained from ATCC (CRL-3216). The packaging virus and the target vector were co-transfected into human embryonic kidney cells HEK293T using lipofectamine 2000 (cell confluency 80–90%). After 48 h of cell culture, the supernatant was collected, containing viral particles after filtration and centrifugation. Virus titers were determined by collecting the virus in the logarithmic growth phase. The lentivirus vector overexpressing PCBP2 was constructed and packaged by Genewiz [28].
Cell transfection and groupingDuring the logarithmic growth phase of cells, digestion with trypsin and trituration were performed to prepare a cell suspension at a concentration of 5 × 104 cells/mL, which was then seeded into 6-well plates at 2 mL per well. Prior to cell grouping, lentivirus particles (multiplicity of infection, MOI = 10, viral titer of 1 × 108 TU/mL) were added to the cell culture medium containing the surfactant polybrene (TR-1003, Sigma-Aldrich, USA) and incubated for 48 h. Stable cell lines were selected with 2 µg/mL puromycin (HY-K1057, Med Chem Express, USA) over a period of 2 weeks. RNA and protein levels were assessed 48 h post-transfection to confirm silencing or overexpression efficiency. The treatment concentrations for CUR and NVs-CUR were both 100 ng/mL.
PC12 cell grouping was conducted as follows: Control group (blank control), H2O2 group (treated with H2O2 for 4 h), CUR group (treated with CUR after H2O2 treatment for 4 h), NVs-CUR group (treated with NVs-CUR after H2O2 treatment for 4 h).
Within the H2O2 group of PC12 cells, further subgroups were designated as follows: (1) sh-NC group (transfected with lentivirus sh-NC), sh-PCBP2 group (transfected with lentivirus sh-PCBP2), oe-NC group (transfected with lentivirus oe-NC), or-PCBP2 group (transfected with lentivirus oe-PCBP2); (2) NVs-CUR + sh-NC group (treated with NVs-CUR and transfected with lentivirus sh-NC simultaneously), NVs-CUR + sh-PCBP2 group (treated with NVs-CUR and transfected with lentivirus sh-PCBP2 simultaneously); (3) sh-NC group (transfected with lentivirus sh-NC), sh-SLC7A11 group (transfected with lentivirus sh-SLC7A11). Among these, the NVs-CUR + sh-PCBP2 group of PC12 cells was further subdivided into: oe-NC group (transfected with lentivirus oe-NC), oe-SLC7A11 group (transfected with lentivirus oe-SLC7A11) [29].
Determination of reduced glutathione (GSH) and oxidized glutathione (GSSG)Cell samples were treated with trypsin, and cell suspensions were prepared by centrifugation to remove debris. The samples were then lysed using a cold freeze–thaw solution. The levels of intracellular GSH and GSSG were measured according to the manufacturer’s instructions using the commercially available GSH and GSSG assay kit (S0053, Beyotime Biotechnology, China). The absorbance of the samples was measured at 450 nm using a plate reader (Infinite200, Tecan, Beijing, China), and quantification was done using a standard curve. Additionally, hypothalamic tissue samples from each group of mice were collected. The tissues were homogenized in lysis buffer, rapidly homogenized using a tissue homogenizer under frozen conditions until fully disrupted, centrifuged at high speed to separate cell debris and undissolved tissue, and the supernatant was collected for GSH content determination using the assay kit [30].
Determination of malondialdehyde (MDA)The levels of MDA in various cell groups and hypothalamic tissues of mice were quantified using the MDA assay kit (S0131S, Beyotime Biotechnology, China) [31].
Determination of Fe2+ contentThe levels of Fe2+ in cells were assessed using the FerroOrange probe (F374, Dojindo, Japan). Pre-treated PC12 cells were seeded on confocal culture dishes, washed with Hank’s balanced salt solution (HBSS, 13150016, Gibco, USA), and then incubated with 1 μM FerroOrange for 30 min. The fluorescence intensity was observed under a confocal laser scanning microscope (LSM780, Zeiss), and the average fluorescence intensity within the cells was measured to evaluate the Fe2+ content. Simultaneously, the iron levels in the cells were determined using an iron assay kit (ab83366, Abcam, UK), following the manufacturer’s instructions [32].
TEM observation of mitochondrial damageMitochondrial damage in PC12 cells was observed using TEM. The samples were fixed overnight at 4 °C in a 25% glutaraldehyde solution (G5882, Sigma-Aldrich, USA), followed by fixation in a 1% osmium tetroxide solution (209104, Sigma-Aldrich, USA) for 1–2 h at room temperature. Dehydration was carried out in a series of graded ethanols (50%, 70%, 80%, 90%, and 95%) at room temperature, followed by treatment with pure acetone (ST1663, BiyunTian Biotechnology, China) and overnight embedding in a pure embedding agent. The embedded samples were then heated at 70 °C overnight to obtain well-embedded specimens. Subsequently, the specimens were sliced to a thickness of 70–90 nm using the UM10 ultramicrotome (Jiangsu Leibo Scientific Instrument Co., Ltd., China). These slices were stained with lead citrate solution (M67147, Shanghai Mayer Biochemical Technology Co., Ltd.) and 2% uranyl acetate saturated aqueous solution (541-09-3, Guangdong Yunxing Biotechnology Co., Ltd.) for 15 min each, enabling observation under the transmission electron microscope [32].
RNA immunoprecipitation (RIP)The EZ-Magna RIP RNA-Binding Protein Immunoprecipitation kit (Merck Millipore, USA) was utilized to investigate the interaction between PCBP2 and SLC7A11 mRNA in PC12 cells. RIP was performed following the manufacturer’s instructions. The general procedure was as follows: initially, the fusion of PCBP2 with a Flag-tag sequence at the DNA level was constructed to generate an expression vector. The constructed Flag-PCBP2 expression vector, along with an empty plasmid vector, was transfected into PC12 cells via lentivirus transfection to induce overexpression within the cells. The cells were harvested and lysed in RIPA lysis buffer. Subsequently, the cell lysates were incubated overnight with IP buffer coated with anti-mouse IgG (ab205718, Abcam, UK) or anti-Flag-PCBP2 (ab251324, Abcam, UK) magnetic beads. Following this, the RNA–protein complexes were washed and incubated with 50 µl of proteinase K (107393, Sigma-Aldrich, USA), and total RNA was extracted using TRIzol reagent for PCR-qPCR analysis [33].
RNA pull-downBiotinylated RNA probes targeting a specific region of SLC7A11 mRNA were prepared using in vitro transcription. Cells were collected and lysed using an RNase inhibitor (HY-K1033, Med Chem Express, USA) and RIPA lysis buffer to isolate RNA and proteins. The biotinylated RNA probes were incubated with the cell lysate, followed by the addition of streptavidin-coated magnetic beads (HY-K0208, Med Chem Express, USA) to capture the biotinylated RNA and its associated proteins. The mixture was incubated at 4 °C for 3 h, then washed twice with cold lysis buffer, three times with low-salt buffer (150 mM NaCl), and once with high-salt buffer (500 mM NaCl). Protein extraction was performed for Western blot analysis to detect the expression of PCBP2 [34].
Analysis of RNA stabilityTo assess the impact of PCBP2 on the stability of SLC7A11 mRNA, we treated cells with Actinomycin D (SBR00013, Sigma-Aldrich, USA) at a concentration of 5 μg/mL. After incubation for designated periods, cell samples were collected at 2, 4, 6, and 8 h. Subsequently, RNA was extracted from each sample, and the expression levels of SLC7A11 were analyzed using RT-qPCR. For detailed methodology, please refer to the following RT-qPCR section [35].
Ethical statementThis study strictly adheres to the relevant ethical principles and regulations regarding animal experiments. All experimental procedures have been approved by the Institutional Animal Care and Use Committee (IACUC) and were approved by the Animal Ethics Committee of General Hospital of Xinjiang Military Command (No. DWLL20200111). All animals are housed and cared for in conditions that meet humane principles and are subjected to experiments with the utmost effort to minimize pain. At the conclusion of the experiments, all mice are euthanized humanely under ether anesthesia.
Establishment and grouping of the mouse modelHealthy male C57BL/6 mice aged 8–12 weeks weighing 20–25 g were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd., China. The mice were individually housed in cages under controlled conditions of 60–65% humidity and temperatures ranging from 22 to 25 ℃, with a 12-h light/dark cycle in a specific pathogen-free (SPF) animal facility. After an acclimatization period of 1 week with standard feeding, the mice were observed for their health status prior to the experiment. The C57BL/6 mice were randomly assigned to the Control group (n = 8) and the Model group (n = 80). The procedure for model establishment involved placing the mice in a specialized constant temperature chamber with a temperature set at 41–43 °C and relative humidity of approximately 40–60% for a duration of 10–15 min, during which the mice had no access to water or food. Following the heat stress, the mice were swiftly transferred to room temperature for rewarming until their body temperature returned to normal (approximately 36.5–37.5 °C). Subsequent to the heat stress and rewarming, the mice underwent close observation to monitor changes in behavior and physiological status, with a record of the survival rate. Two weeks after the heat stress treatment, the degree of damage to the hypothalamic neuroaxis was assessed through neurological deficit scoring and histological examination (H&E staining and Nissl staining) to confirm the successful establishment of the model [36].
The Model group mice were further randomly divided into the following subgroups: (1) CUR group, NVs-CUR group; (2) NVs + DMSO group, NVs-CUR + DMSO group, and NVs-CUR + Erastin group; (3) NVs + sh-NC + oe-NC group, NVs-CUR + sh-NC + oe-NC group, NVs-CUR + sh-PCBP2 + oe-NC group, and NVs-CUR + sh-PCBP2 + oe-SLC7A11 group, each consisting of 8 mice. Prior to modeling, each mouse received a single tail vein injection of lentivirus at a dose of 6.4 × 106 TU in a volume of 20 μL. After 12 h of heat stress, CUR and NVs-CUR (2 × 109) were administered intravenously, with a dose of 30 mg/kg/day for CUR, both given continuously for two weeks [37]. After heat stress treatment, the ferroptosis activator Erastin (HY-15763, Med Chem Express, USA) was administered via intravenous injection at a dose of 30 mg/kg once daily for two weeks [38].
In vivo fluorescence imaging of NVs-CURTo assess the biodistribution of CUR and NVs-CUR in the mouse brain, FITC-labeled CUR (Q-0127482, QIYUE Biology, China) and NVs-CUR were administered 2 h prior to injection. Subsequently, at 6 and 24 h post-injection, the in vivo fluorescence signals of the mice were detected using the IVIS Lumina III live imaging system (PerkinElmer, CLS136334). Prior to imaging, the mice were briefly anesthetized to ensure their immobility throughout the procedure [39].
Detection of neuronal uptake of NVs-CUR by flow cytometryFollowing in vivo fluorescent imaging, neuronal distribution of NVs-CUR in the hypothalamic tissue of Model mice was analyzed using FITC-labeled anti-NeuN antibody (ab177487, 1:1000, Abcam, UK) as a surface marker, with neuronal sorting conducted via the Cytek Aurora Flow cytometer. Subsequently, the percentage of cells uptaking DiR-labeled NVs and NVs-CUR was determined [39].
Neurological function deficit scoringBased on a previously reported scoring system [40], we assessed the neural damage in each group of mice before and after modeling at 1, 3, 7, and 14 days, which involved motor, sensory, and balance tests. The scoring ranged from 0 to 10, with higher scores indicating more severe neural damage. Exclusion criteria were: (a) death within the specified time frame; (b) obtaining a score of 0 at any given time.
The wire-hanging test was conducted to evaluate the mice’s motor coordination ability and muscle strength. The procedure involved preparing a wire with a diameter of approximately 2–3 mm. Prior to the formal experiment, the mice were familiarized with the wire through brief training without scoring. They were then gently hung by their front paws on the center of the wire, and the timing started from when they grasped the wire until they dropped. Each mouse underwent the test three times with intervals of no less than 15 min. The scoring was based on the duration the mice hung, with shorter durations indicating more severe motor impairment [41].
The bilateral tactile stimulation test was performed to evaluate the mice’s responsiveness to tactile stimuli. The mice were placed in the experimental cage and allowed to acclimate for approximately 5 min before lightly touching their flanks alternately with a brush or cotton swab to avoid eliciting a stress response. Responses such as turning, licking, or evading touch were recorded and graded based on the intensity of the mouse’s reaction to the tactile stimuli, with weaker responses indicating more severe sensory impairment [42].
The balance beam walking test was conducted to assess the mice’s balance and coordination. Prior to the formal experiment, the mice were allowed to walk freely on the balance beam to adapt to the environment. Subsequently, each mouse was placed at one end of the beam, and the time taken to walk to the other end was recorded. The stability of the mice during walking, including swaying, pausing, or falling, was observed. Scores were assigned based on the completion speed of the test and the stability during walking, where slower speed and poorer stability indicated a more severe impairment in balance ability [43].
H&E stainingAt the conclusion of the experiment, all mice were euthanized by inhalation of isoflurane (1% O2). The hypothalamic tissues were prepared for fixation by perfusion and then washed three times for 12 h each with PBS, followed by immersion in a 30% sucrose solution at room temperature for 24 h. The tissues were embedded in a chilled embedding medium and immediately frozen. The hypothalamic tissues were then sliced into 7 µm sections using a cryostat (Leica Microsystems) for further analysis. These prepared sections were stained in hematoxylin–eosin solution (H8070, Solarbio, Beijing, China) for typically 5 to 10 min at room temperature. Subsequently, the sections were rinsed with distilled water, dehydrated in 95% ethanol, and placed in an eosin staining solution (G1100, Solarbio, Beijing, China) for 5 to 10 min. Standard dehydration, clearing, and mounting procedures were followed [44].
Nissl stainingThe frozen sections, as described previously, were rinsed in distilled water for 2 min to a thickness of 7 μm. Subsequently, the sections were stained with Nissl staining solution (C0117, Biyun Technologies, China) at 37 °C for 10 min, followed by replacement with fresh distilled water and a 2-min wash. Next, they were briefly immersed in 95% ethanol for 5 s, followed by two washes in 70% ethanol every 5 s. The stained sections were then visualized and scanned using the Pannoramic MIDI CaseViewer 2.0 system (3DHISTECH Ltd.) [36].
Immunofluorescence stainingFor the immunofluorescence staining of the hypothalamic tissue, we conducted DAPI and NeuN staining to acquire representative confocal immunofluorescence images. The procedure involved the following steps: tissue sections were subjected to antigen retrieval treatment, delineated with a hydrophobic barrier using a histology pen to outline the tissue surrounding the region of interest, and then immersed the slices in a 3% H2O2 solution to block endogenous peroxidase activity. Subsequently, the sections were blocked with serum or BSA and incubated overnight at 4 °C with rabbit monoclonal anti-NeuN antibody (ab190565, 1:50, Abcam, UK). Following this, incubation with DAPI (ab104139, 1: 1000, Abcam, UK) was performed [45].
For the cellular immunofluorescence staining, cells were washed with ice-cold PBS and fixed with 4% paraformaldehyde (P885233, Macklin, Shanghai, China) for 15–30 min. Subsequently, cells were permeabilized with 0.1% Triton X-100 (L885651, Macklin, Shanghai, China) for 15 min to penetrate the cell membrane. After two washes with PBS, cells were incubated in PBS with 15% FBS at room temperature for 1 h. The cells were then incubated overnight at 4 ℃ with rabbit polyclonal anti-PCBP2 antibody (ab184962, 1:250, Abcam, UK). On the following day, sections were washed with PBS, incubated with goat anti-rabbit secondary antibody (ab150115, 1:200, Abcam, UK) at room temperature for 1 h, rinsed with PBS, and stained with DAPI (ab104139, 1:1000, Abcam, UK) for 5 min to label the nuclei. Excess DAPI was removed by washing the slides three times with PBS for 5 min each. Subsequently, fluorescence imaging was performed using a Zeiss Observer Z1 microscope (Germany). Five random fields of view were chosen, the proportion of PCBP2-positive cells was quantified in each field, and the mean value was calculated [46].
ELISA assayThe hypothalamic tissue was washed in PBS and homogenized using a homogenizer in a centrifuge tube containing an appropriate amount of protein extraction buffer, followed by centrifugation of the homogenate and collection of the supernatant. Mouse ELISA kits for IL-1β (PI301), IL-6 (PI326), and TNF-α (PT512) from Biotech Inc., China were selected for analysis and performed according to the manufacturer’s instructions [47].
RT-qPCRTotal RNA was extracted using the Trizol reagent kit (T9424, Sigma-Aldrich, USA). The quality and concentration of RNA were assessed using UV–visible spectrophotometry (ND-1000, Nanodrop, Thermo Fisher, USA). Reverse transcription was performed following the PrimeScript™ RT-qPCR kit (RR086A, TaKaRa, Mountain View, USA). Real-time quantitative reverse transcription polymerase chain reaction (RT-qPCR) was carried out on the LightCycler 480 system (Roche Diagnostics, Pleasanton, USA) using SYBR Premix Ex Taq™ (DRR820A, TaKaRa, Japan). The housekeeping gene GAPDH was used as a reference for mRNA. The primer sequences were designed and provided by Shanghai General Biotech Co., Ltd. Refer to Table S2 for primer sequences. The fold change in target gene expression between the experimental and control groups was calculated using the 2−ΔΔCt method, where ΔΔCT = ΔCt experimental group—ΔCt control group, and ΔCt = Ct target gene—Ct housekeeping gene [48].
Western blotCellular or tissue total protein was extracted using the efficient RIPA lysis buffer following the manufacturer’s instructions. After 15 min of lysis at 4 °C and centrifugation at 12,000 × g for 15 min, the supernatant was collected, and the protein concentration of each sample was determined using the BCA assay kit (20201ES76, Yisheng Biotechnology Co., Ltd., Shanghai, China). Quantification was performed based on different concentrations. Proteins were separated by polyacrylamide gel electrophoresis and transferred to a PVDF membrane using the wet transfer method. The membrane was blocked with 5% BSA at room temperature for 1 h. Primary antibodies were then incubated overnight at 4 °C (antibody information in Table S3). Subsequently, the membrane was washed with TBST for 5 min × 3 times, followed by incubation with HRP-conjugated goat anti-rabbit IgG (ab205718, 1:20,000, Abcam, UK) dilution at room temperature for 1 h. After washing the membrane with TBST for 5 min × 3 times, the membrane was developed using a chromogenic substrate. Protein quantification analysis was performed using ImageJ software (v1.48, National Institutes of Health) by evaluating the ratio of the grayscale values of each protein to the internal control GAPDH [49]. The experiment was repeated three times.
Statistical analysisAll experiments were independently carried out at least three times, and the data were presented as mean ± standard deviation (SD). Differences between groups were assessed using independent samples t-test or one-way analysis of variance (ANOVA). If the ANOVA results indicated significant differences, further Tukey’s Honestly Significant Difference (HSD) post hoc tests were conducted to compare differences among groups. For non-normally distributed or inhomogeneous variance data, the Mann–Whitney U test or Kruskal–Wallis H test was utilized. All statistical analyses were performed using GraphPad Prism 8.0 software. A significance level of P < 0.05 was considered statistically significant [50].
留言 (0)