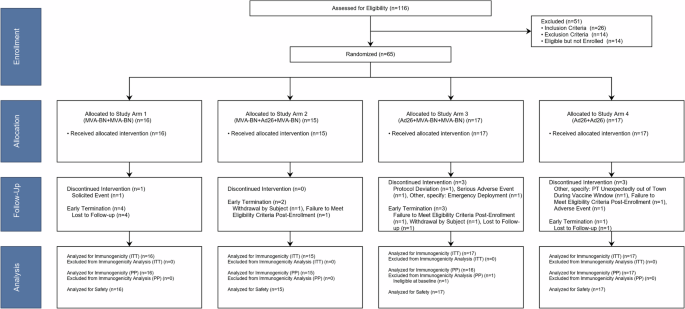
Trial design and participants
This study was reviewed and approved by the Emory University Institutional Review Board (IRB00087894) and all participants provided written informed consent. The NCT number was NCT02891980, “A Safety Trial to Test MVA-BN(R)-Filo and Ad26.ZEBOV Vaccines in Healthy Volunteers,” registered September 1, 2016. This was a Phase 1, double-blinded randomized trial to evaluate the safety and immunogenicity of two heterologous and two homologous prime-boost regimens using MVA-BN®-Filo and Ad26.ZEBOV administered in different sequences at Days 1 and 29 in healthy adult participants aged 18–45 years. The two heterologous prime-boost arms (Study Arms 2 and 3) also received a late MVA-BN®-Filo boost at Day 366. Participants and study staff were blinded to a participant’s study vaccine assignment within study vaccination schedule (e.g., enrollment into Study Arm 1 or 4 versus 2 or 3 was known). Participants randomized to Study Arms 1 and 4 (2-dose schedule) were followed through ~12 months after the first study vaccination, while those randomized to Groups 2 and 3 (3-dose schedule) continued follow-up through ~18 months after the first study vaccination (6 months after the third study vaccination). This study planned for 60 participants to be randomized 1:1:1:1 to one of four Study Arms (i.e., 15 per arm), but participants who did not receive the second study product dose were replaced.
A thorough medical history, physical examination, an electrocardiogram (ECG), and laboratory tests were obtained at screening. Enrollment criteria included healthy adult participants aged ≥18 to ≤45 years without a history of Ebola disease, exposure to Ebola or prior vaccination with an Ebola vaccine, and receipt of any Ad26-based vaccine or vaccinia (smallpox)-based vaccine. See Supplementary Methods for additional information. Participants were followed as per their randomization assignment.
Study product and administration
MVA-BN®-Filo dose was administered at 1 × 108 international units (IU) per dose as a 0.5 mL intramuscular (IM) injection in the deltoid, and the Ad26.ZEBOV dose was administered at 5 × 1010 virus particles (VP) per dose as a 0.5 mL IM injection in the deltoid. The Adenovirus 26-vectored vaccine (Ad26.ZEBOV) expresses the EBOV glycoprotein, while the modified vaccinia Ankara-vectored vaccine expresses glycoproteins from EBOV, SUDV, and MARV, and the nucleoprotein from Taï Forest virus (MVA-BN-Filo).
Data collection
The occurrence of solicited injection site and systemic reactogenicity events was measured from the time of study vaccination through Day 8 after each vaccination. Unsolicited AEs were collected from vaccination through Day 29 after each vaccination. SAEs, AEs of special interest (AESIs), and AEs related to blood draws were collected from enrollment through the end of the study.
Assessment of responses
The study included phlebotomy for transcriptomics prior to each dose of vaccine (Day 1), and Days 2, 4, 8, 15, and 29 after each dose of vaccine. Plasmablast responses were obtained prior to each vaccination (Day 1) and Day 8 after each dose of vaccine. Serology and cellular responses were obtained prior to each dose of vaccine (Day 1), and at Days 8, 15, and 29 after each vaccination. Durability data was obtained at 6 and 11 months after the second dose in all participants and at 6 months after the third dose in Study Arms 2 and 3.
Binding and neutralizing antibody responses
Antibody responses were assessed using anti-EBOV-GP (Kikwit) IgG enzyme-linked immunosorbent assay (ELISA) (measured in ELISA Units (EU) per mL) at Battelle, Columbus, Ohio as previously described50. The EBOV GP (Makona) pseudovirus viral neutralization assay (VNA) (measured in IC50 titers) was performed at Monogram Biosciences, Inc, San Francisco, CA as previously described28. Briefly, EBOV GP pseudoviruses were generated using a replication defective retroviral vector containing a luciferase reporter gene. Pseudoviruses were incubated with serial dilutions of participant sera and used to infect humab embryonic kidney (HEK) 293 cell cultures. Luciferase activity was measured at ~72 h postviral inoculation to determine serum neutralizing antibody titers. These were summarized on a per-visit, fold change (relative to pre-vaccination), and peak response level following first, second, and third vaccination (as applicable).
Antibody-dependent cell-mediated cytotoxicity (ADCC) responses
To measure ADCC responses, dual-reporter target cell lines with tetracycline-inducible expression of green fluorescent protein, luciferase, and individual filovirus GPs (EBOV, SUDV, MARV, TAFV) were generated as previously described for other pathogens (Supplementary Methods)32. The effector cells for the assay were CD16-176V-NK-92 cells (obtained from Fox Chase Cancer Center, Philadelphia, PA) which have high surface expression of CD16. These cells were maintained in Mylocult media supplemented with 200 IU/mL of recombinant human IL-2 (R&D Systems, Minneapolis, MN).
To perform ADCC assays, serum samples were serially diluted 1:3 with a starting dilution of 1:20. Serum was mixed with target cells and NK effector cells with an effector:target (E:T) ratio of 2:1 and incubated for 4 h at 37 °C. Following incubation, Britelite Plus luciferase reagent (PerkinElmer) was added and Relative luminescence units (RLUs) were read on a luminometer (TopCount NXT Luminescence Counter). To calculate the percent ADCC, we determined the percent lysis of target cells using the following formula: ADCC (%) = [RLU × (no antibody) − RLU (with antibody)]/RLU (no antibody)] × 100. The endpoint ADCC titer was defined as the highest serum dilution at which >24% ADCC was observed for a given sample.
Plasmablast responses
To measure total and Ebola-specific (EBOV GP, SUDV GP, MARV GP) plasmablast responses, an ELISpot assay was implemented as previously described8,51. Briefly, 96-well ELISpot plates (EMD Millipore, Billerica, MA) were coated with 100 μl of either polyclonal coating antibodies (goat anti-human IgA + IgG + IgM (H + L), 10 μg/ml, Jackson Immuno Research, West Grove, PA), or with 3.0 μg/ml of recombinant EBOV GP, SUDV GP, or MARV GP (IBT Bioservices, Gaithersburg, MD) and incubated at 4 °C overnight. Plates were blocked with RPMI media (Cellgro, Manassas, VA) supplemented with 10% heat inactivated fetal bovine serum (Sigma-Aldrich, St. Louis, MO) for 2 h at 37 °C. Following washing, freshly isolated PBMCs (5 × 105 cells per well), were added to the wells and plates incubated overnight at 37 °C in 5% CO2. Following incubation, plates were washed four times with phosphate buffered saline (PBS) and four times with PBS supplemented with 0.05% Tween 20 (PBS-T). Subsequently, 100 μl of 1:1000 diluted biotin-conjugated donkey anti-human IgG and goat anti-human IgA and IgM (Jackson) were added to each well and plates incubated at room temperature for 2 h. Following another washing with PBS-T/PBS, plates were incubated with 100 μl of 1:1000 diluted horseradish peroxidase conjugated avidin D (Vector Laboratories, Burlingame, CA) for 1 h at RT. Plates were thoroughly washed and the reaction was developed with the addition of 100 μl/well of AEC Substrate (BD Biosciences, San Diego, CA). Reactions were allowed to develop at RT and stopped, when spots became visible in the wells with highest density of the cells (generally 5–8 min), by discarding the reaction mix and washing the plates under running cold water. Plates were allowed to dry and were read in an ELISpot reader (CTL, Shaker Height, OH). Final data was calculated as antibody secreting cells (ASC) per million PBMC.
Cellular responses by Intracellular cytokine staining (ICS)
ICS of PBMCs collected at various time points (pre-vaccination and post-vaccination) were assessed for the production of interferon-γ (IFN-γ), tumor necrosis factor α (TNF-α), and interleukin-2 (IL-2) from T cells stimulated with antigen-specific peptide pools. Customized peptide pools of Ebola virus Glycoprotein (EBOV GP, Zaire Mayinga strain, GenBank # AF086833.2), Sudan virus Glycoprotein (SUDV GP, Sudan Gulu strain, Genbank # AY729654), Marburgvirus Glycoprotein (MARV GP, Marburgvirus Musoke strain, GenBank # DQ217792.1) and Taï Forest virus Nucleoprotein (TAF NP, Taï Forest ebolavirus, GenBank # KU182910.1), were synthesized by Genescript. Each pool had 40 peptides, which are each 15 amino acids with overlapping by 11 amino acids.
Detailed methods are described in Supplementary Methods. Briefly, PBMCs were thawed and incubated with peptide pools for 6 h in the presence of Brefeldin A (BD Bioscience, Cat # 555028). The final peptide concentration was 2 μg/mL for each peptide. Stimulated cells with Staphylococcal Enterotoxin B (SEB) or DMSO (peptide diluent) was used as a positive or negative control, respectively. Then, cells were washed and stained with viability dye (Zombie aqua, BioLegend, Cat # 423102). Next, cells were surface stained with BV605-CD3 (HIT3a, BD Bioscience, Cat # 5647120), eFluor 450-CD4 (OKT-4, eBioscience, Cat# 48-0048-42), APC-Cy7-CD8 (RPA-T8, BD Bioscience, Cat # 557760), PE-CF594-CCR7 (150503, BD Bioscience, Cat # 562381), and PE-Cy7-CD45RA (HI100, BD Bioscience, Cat# 560675) for 30 minites at 4 °C, followed by permeabilization using Cytofix/Cytoperm (BD Bioscience, Cat # 555028). To quantify cytokines, PE-IFN-γ (4S.B3, BioLegend, Cat # 502509), FITC-IL-2 (MQ1-17H12, BioLegend, Cat # 500304), and Alexa 647-TNF-α (MAb11, BioLegend, Cat # 502916) were used. Fluorescence intensity was measured using an LSRII cytometer (BD Biosciences) and a Symphony A5 (BD Biosciences). Data analysis was performed using FlowJo software (Tree Star). Samples were classified as positive or negative for a given cytokine based on pre-defined cut-off values established by the laboratory.
Transcriptomics
For transcriptomics, a single aliquot per time point and subject was analyzed. PBMCs were lysed in Buffer RLT (Qiagen) containing 1% b-mercaptoethanol and stored at −70 °C for later extraction. RNA was isolated from each sample using the RNeasy Mini kit (Qiagen) with on-column DNase digestion at the Emory Transcriptomics Core. RNA quality was assessed using an Agilent Bioanalyzer and 500 ng of total RNA was used as input for library preparation using the TruSeq mRNA Stranded Kit (Illumina) according to the manufacturer’s instructions. As an internal control, an ERCC ExFold RNA Spike-In Mix (ThermoFisher) was also added to each sample well according to manufacturer instructions. Libraries were validated by capillary electrophoresis on an Agilent 4200 TapeStation, pooled at equimolar concentrations, and sequenced on an Illumina HiSeq3000 at 100SR, yielding 25–30 million reads per sample. All reads were extracted using standard Illumina sequencing demultiplexing (bcl2fastq) with no processing, filtering, or cleaning.
Statistical analysesSample size
The sample size for this study was selected to obtain preliminary estimates of vaccine safety and immunogenicity in a time sensitive manner. The study was not designed to test a specific null hypothesis.
ELISA and neutralizing antibody analyses
For each study arm and analysis population, titers or ELISA units/mL results and fold changes for applicable study visits were summarized by tabulating the number of observations, geometric mean, and 95% CI of the geometric mean (based on Student’s t-distribution), geometric standard deviation, median, first and third quartile, minimum, and maximum. In addition, peak titers were compared in a pairwise fashion between the four study arms using a two-sided Welch’s t-test.
The estimate of the population mean difference in log2 peak value following each vaccination and the associated two-sided 95% confidence intervals (CI) for each comparison were calculated using the Welch–Satterthwaite method. The difference was also presented on the original scale representing the ratio of the geometric means and associated 95% CI. To assess if the third vaccine dose for Study Arms 2 and 3 resulted in statistically significantly increased or decreased peak values following third vaccination, a two-sided paired t-test was used to compare mean log2 peak values post third and post second vaccination within Study Arms 2 and 3.
Antibody-dependent cellular cytotoxicity analyses
Prior to analysis, ADCC titers that were negative (titer < 1:20) were assigned a titer of one half the lowest dilution (i.e., 1:10). Post-vaccination samples for participants that were negative, i.e., had a titer below 1:20 at baseline were classified as positive if the respective post-vaccination day sample was positive, i.e., had a titer greater than or equal to 1:20. Post-vaccination samples for participants that were positive at baseline were classified as positive if the respective post-vaccination showed ≥4-fold increase compared to pre-first vaccination (Day 1). For each analysis population (ITT and PP), ADCC titer fold change compared to pre-vaccination was summarized using descriptive statistics (Minimum, Q1, Median, 95% CI of the median, Q3, and Maximum) by study arm and post-vaccination time point. The non-parametric bootstrap method with 1,000 bootstrap replicates was used to obtain 95% CIs of the median for each treatment arm and study visit combination. In addition, positive response on a per-visit basis was summarized using n/NNM (%) and associated Clopper-Pearson 95% CIs where n represented the number of participants who had a positive response at the visit and NNM represented the number of participants for that particular study visit and treatment combination with non-missing laboratory results.
For each participant, peak ADCC fold change response following first vaccination was defined as the maximum post-first vaccination fold change (for Days 15 and 29 relative to post-first vaccination) compared to pre-first vaccination (Day 1). For each participant, peak ADCC fold change response following second vaccination was defined as the maximum fold change post-second vaccination (for Days 15, 29, and 181 post-second vaccination and Day 366 post-first vaccination) compared to pre-first vaccination (Day 1). For each participant in Study Arms 2 and 3, peak ADCC fold change response following third vaccination was defined as the maximum fold change post-third vaccination (for Days 15, 29, and 181 post-third vaccination) compared to pre-first vaccination (Day 1). A two-sided Wilcoxon Rank-Sum test (normal approximation) was applied to assess difference in peak ADCC titer fold change compared to pre-vaccination between treatment arms for each analysis population and dose. Results were summarized using the number of participants in each arm, medians of the peak fold change response for each arm, the Wilcoxon Rank-Sum test statistic and p value. In addition, peak positive response (the number of participants who had a positive response at any point in the postvaccination period) was compared in a pairwise fashion between study arms using a two-sided Fisher’s exact test. Results were summarized using n/NNM (%), Odds Ratio, and P value where n represented the number of participants who had a positive response and NNM represented the number of participants for that particular peak response and treatment comparison with non-missing laboratory results.
Cell-mediated immunity treatment arm comparisons
Additional summaries of this data for the exploratory endpoint included the comparison of the peak difference in the percentage of activated T cells post-first, post-second, and post-third vaccination. A two-sided Wilcoxon Rank-Sum test (normal approximation) was used to assess the statistical significance of applicable pairwise study arm comparisons. Results were summarized using the number of participants in each arm, medians of the peak response for each arm, the Wilcoxon Rank-Sum test statistic and p value. In addition, peak positive response (the number of participants who had a positive response at any point in the post-vaccination period) was compared in a pairwise fashion between study arms using a two-sided Fisher’s exact test.
Results were summarized using n/NNM (%), Odds Ratio, and p value where NNM represented the number of participants for that particular peak response and treatment comparison with non-missing laboratory results.
Plasmablast analysis
Prior to analysis, technical replicates were aggregated using the mean. The lower limit of detection (LOD) was specified as 7 cells per million. The upper limit of quantification (ULOQ) was specified as 85 cells per million. Results for cell populations that were below the LOD were imputed using 0.5 x LOD = 3.5 cells per million. Cell populations too numerous to count were imputed using the 2 × ULOQ = 170 cells per million. Analysis was performed for the exploratory plasmablast analysis population. The number of secreting plasmablast cells per million and fold change relative to pre-vaccination were summarized for each antibody type and Ebolavirus GP antigen, applicable timepoint, and study arm using the minimum, 25th percentile (Q1), median, 95% CI of the median, 75th percentile (Q3), and maximum. The non parametric bootstrap method with 1000 bootstrap replicates was used to obtain 95% CIs of the median. For each dose, antibody type, and Ebolavirus GP antigen, a two-sided Wilcoxon Rank-Sum test was performed to assess the difference in peak fold change in secreting plasmablast cells per million compared to pre-vaccination between treatment arms. Correlations between the change in the number of secreting plasmablasts and peak humoral and cellular assay results were assessed using Spearman correlation.
Gene expression biomarkers predicting peak VNA titer
Regularized linear regression models were fit to determine gene expression fold-change responses that best predict peak virus neutralization assay (EBOV GP VNA) antibody titer post-second vaccination using the glmnet R package (Version 3.0.2) [17]. To avoid overfitting (n « # genes and collinearity among genes) and to facilitate variable selection, an elastic net regularization step (combination of L1 Lasso and L2 ridge penalization, a = 0.5) was applied. The analysis was carried out across treatment arms. Six-fold stratified cross validation (sets of 10 participants) was used to determine the optimum regularization parameter that minimizes the model mean squared error and peak EBOV GP VNA titer was utilized for stratifying participants by antibody response using deciles. In both cases, the input gene set was based on log2 fold change in LCPM for DE genes that were identified using binomial models as implemented in edgeR for any timepoint and comparison (within and between study arm FDR < 0.05, FC ≥ 1.5). Regularized linear regression models were fit for Days 2, 4, 8, 15, 29 (relative change compare to first vaccination) and Days 30, 32, 36 (relative change compared to second vaccination).


留言 (0)