
記住我

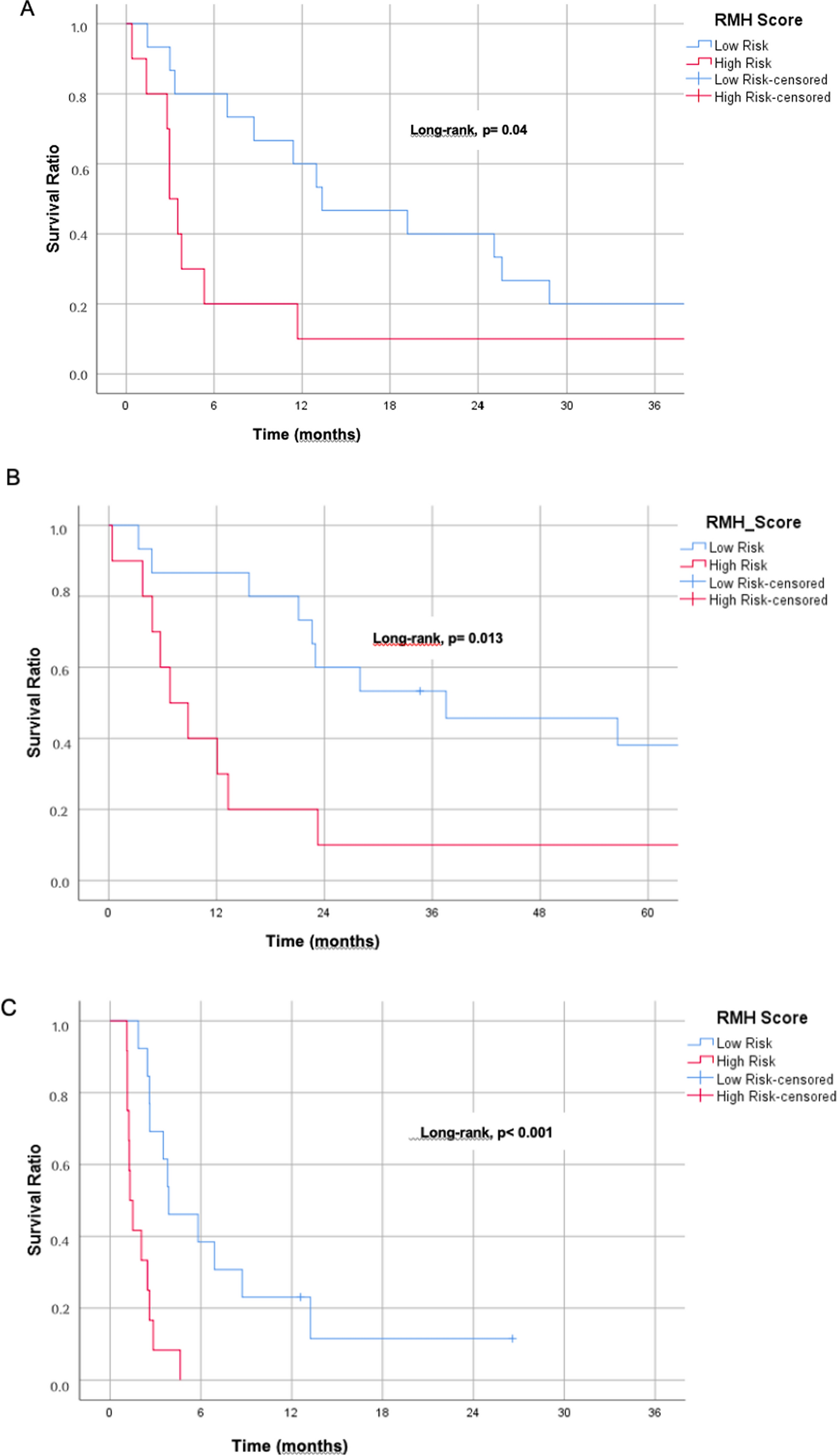



CYP19A1 showing physical interactions (77.64%) with CYP19A1, PBX1, NFIC, HSD3B1, HSD3B2, HSD17B3, PTPA, HSD17B1, RC3H2 and FOXL2 (showing in yellow colour lines). CYP17A1 showed co-expression (8.01%) except HSD17B1, RC3H2, and CEBPD. And CYP19A1 with all except ID3, PTPA, CPB1, NFIC and POR. Showing predicted (5.37%) with CYP21A2, POR, CYP11A1 and CYP2E1. Colocalization (3.63%) with all except HSD17B1, RC3H2, and CEBPD. Genetic interactions (2.87%) with all except HSD17B1, RC3H2, and CEBPD.
By employing Computational analysis, we aimed to provide a comprehensive understanding of the potential of Diosmetin as a CYP inhibitor in prostate cancer therapy (Fig. 1). The findings from this in silico investigation can serve as a basis for future experimental analysis to validate and expand upon the computational results. Furthermore, successfully identifying Diosmetin as a promising lead compound could pave the way for developing novel CYP-targeted therapies against prostate cancer, offering new treatment options for patients facing this challenging disease. Figure 1 shows the association of genes with CYP17A1 and CYP19A1.
Fig. 1
The figure shows physical interaction of CYP19A1 protein
2.1 Virtual screening and molecular docking analysisThe study's findings demonstrate that Diosmetin has a remarkable binding energy of − 11.261 kcal/mol, surpassing that of CYP17A1 and CYP19A1 with a binding energy of − 11.145 kcal/mol (as shown in Figs. 2 and 3). This difference in binding energy implies that Diosmetin may have anti-cancer properties, making it an attractive choice for treating prostate cancer. Furthermore, Diosmetin has favourable drug-like characteristics and pharmacokinetic properties, enhancing its therapeutic potential. This in silico analysis provides a solid framework for subsequent inquiry in both in vitro and in vivo environments. These subsequent studies will shed insight into its varied actions and lay the groundwork for leveraging its bioactive potential against cancer illnesses.
Fig. 2
Showing 2D interaction with A CYP19A1 (− 11.145 kcal/mol) and B CYP17A1 (− 11.261 kcal/mol) and Diosmetin
Fig. 3
Depicts the 3D interactions with A CYP19A1 and B CYP17A1
Table 1 presents a list of selected CYP inhibitors along with their corresponding molecular docking scores (in kcal/mol), which reflect the strength of their binding to various CYP enzymes. Molecular docking is a computational method used to predict how well a small molecule (like a drug or inhibitor) binds to a target enzyme. The docking score is a key indicator of this binding affinity, with more negative values suggesting stronger interactions. For instance, CYP17A1 has a docking score of − 11.3 kcal/mol, indicating a strong binding affinity with the inhibitor. In contrast, CYP19A1 shows a positive docking score of 11.2 kcal/mol, suggesting weak or negligible binding with the inhibitor. Other enzymes, such as CYP1A2, CYP2C9, CYP2C19, CYP3A4, and CYP2D6, show moderate to strong interactions, with docking scores ranging from − 8.3 kcal/mol to − 9.1 kcal/mol. These results provide insight into how effectively these inhibitors interact with the listed CYP enzymes, which could be important for applications in drug metabolism and understanding potential drug interactions. Overall, the table highlights the varying degrees of inhibitor binding to different CYP enzymes, helping to evaluate the inhibitors' potential effectiveness in modulating enzyme activity.
Table 1 List of selected CYP inhibitors docking scores based on molecular docking score (kcal/mol)Furthermore, Diosmetin has favourable drug like characteristics and pharmacokinetic properties, enhancing its therapeutic potential. This in silico analysis provides a solid framework for subsequent inquiry in others environments (Table 2). These subsequent studies will shed insight into its varied actions and lay the groundwork for leveraging its bioactive potential against cancer illnesses.
Table 2 ADME properties of Diosmetin2.2 DFT analysisDensity Functional Theory (DFT) calculations were systematically conducted utilizing the Schrödinger framework to meticulously investigate the molecular geometry and the electron distribution within the solid structure, as illustrated in Fig. 5. The employment of DFT, a pivotal and esteemed computational approach, facilitates an in-depth exploration of the intricate interplay between the geometric attributes and electronic properties of chemical entities. In this study, we present a comprehensive suite of DFT computations, encompassing elaborate analyses such as electronic spectra investigations, scrutiny of Highest Occupied Molecular Orbital (HOMO) to Lowest Unoccupied Molecular Orbital (LUMO) energies accompanied by an assortment of chemical reactivity parameters. These computational pursuits collectively contribute to a profound understanding of the intricate nuances governing the molecular architecture and electronic disposition within the studied system (Fig. 4).
Fig. 4
Shows the HOMO–LUMO of Diosmetin and the accompanying transitioning energies. The energy gap between HOMO–LUMO is 0.154591 and 0.134636 cyp17a1 and cyp19a1 respectively
2.3 ADME analysisADME prediction is critical in drug discovery and development to forecast a possible therapeutic molecule's in vivo pharmacokinetics. Along with molecular docking studies, ADME evaluations are undertaken to classify a pharmaceutical molecule's safety and efficacy. Tables 2 and 3 show the pharmacological properties.
Table 3 Drug-likeness properties of DiosmetinThe investigated chemicals' levels fall within a permissible range in the current investigation. Because the native Diosmetin complex sits between these values, it is easily permeable. The results revealed that the natural chemical Diosmetin followed and did not violate Lipinski's RO5 compared to the screened compounds from Zinc databases. In humans, Diosmetin has outstanding oral absorption and acceptable pharmacokinetic properties. This Diosmetin compound ought to function as an active pharmaceutical molecule and have a high bioavailability as a consequence.
2.4 BOILED egg plot analysisFailures in minor molecule effectiveness and toxicity are brought on by unfavourable pharmacokinetic characteristics and ADME prediction. The Diosmetin has two advantageous pharmacokinetic characteristics: Gastrointestinal Absorption and Blood Blood–Brain Barrier, as illustrated in the Boiled Egg plot Fig. 6. The white zone of the egg, not the yellow zone, indicates the high gastrointestinal absorption rate of this phytochemical (Fig. 5). As a consequence, the brain cannot pass through this Diosmetin molecule. In our research, Diosmetin was found to be the most potential agent to treat Prostate Cancer. But to confirm the results of this work, further studies are required.
Fig. 5
Depicts the Boiled egg graph, which shows Diosmetin's gastro and BBB activities
2.5 Molecular dynamic simulationStability of the CYP17A1 and CYP19A1 proteins with Diosmetin ligand. Figure 7 displays the Diosmetin-CYP17A1 and CYP19A1 protein complex's residual mean square deviation (RMSD) during a 200-ns molecular simulation. The early simulation stage between 0 and 15 ns may be brought on by the CYP17A1 and CYP19A1 proteins' activation loop movements. Beyond the first stage of the molecular dynamics simulation, however, there are either no or extremely few variations (Fig. 6).
Fig. 6
RMSD of the complex with A CYP19A1 and B CYP17A1. The complex is stable throughout the 200 ns simulation between 1.5–2.8 Å (CYP19A1) and 2.8 Å (CYP17A1)
RMSF investigates the activation loop's flexibility and the complex's RMSF map, the main peaks of variance between residues are more than 0.1 nm. These residues are not found in docking experiments. As a consequence, the fluctuation has no effect on protein chemical affinity to Diosmetin. At less than 1 nm, the remaining residues are all stable (Fig. 7).
Fig. 7
RMSF of the complex with A CYP19A1 and B CYP17A1
One of the most important components of the protein–ligand complex is the hydrogen bond. The Desmond package is used to detect the hydrogen bond between Diosmetin and the CYP17A1 and CYP19A1 proteins, as illustrated in Fig. 8A. This interaction can stabilise Diosmetin at the site of contact. Consequently, a hydrogen bonding interaction between Diosmetin and the CYP17A1 and CYP19A1 proteins is consistent with the docking study findings (Fig. 8B).
Fig. 8
A The interactions contacts of CYP19A1 and B CYP17A1 with Diosmetin are shown from 200 ns simulation time. Left side represented the protein-ligands contact and right side represented the ligand–protein contacts
2.6 Ligand torsion profile and propertiesThe ligand torsions chart and other attributes show how each rotatable bond (RB) in the ligand [13] experienced a conformational shift over the simulation trajectory (0.00 to 200 ns) [14]. The top panel displays a picture of a two-dimensional ligand with coloured, rotatable bonds. A dial plot and correspondingly coloured bar graphs are shown with each rotatable bond torsion. Dial (or radial) charts show the evolution of the torsion over the simulation. The simulation's starting point, located in the centre of the radial map, is the origin of the temporal progression. The probability density of torsion is used in the bar graphs to summarise the data on the dial plots (Figs. 9).
Fig. 9
Demonstrates the ligand torsion profiles in 2D and 3D for A CYP19A1 and B CYP17A1
Additionally, we analyzed the hydrogen bonds, solvent-accessible surface area (SASA), radius of gyration (rGyr), and MolSA for the selected complexes. SASA, rGyr, and MolSA analyses of CYP19A1 and CYP17A1proteins revealed that Diosmetin exhibited significant stability and most suitable targets for prostate cancer (Fig. 10A and B).
Fig. 10
Demonstrates the RMSD, Intra Hydrogen bond, radius of gyration, MolSA, PSA, and SASA, values connected to A CYP19A1 and B CYP17A1
2.7 Protein secondary structureBeta- and alpha-strands are tracked throughout the simulation as examples of protein secondary structural elements (SSE). The SSE distribution throughout the protein structure is shown in the image above by residue index. The graphic below summarises the SSE component for each trajectory frame throughout the simulation, whereas the bottom figure tracks the SSE assignment of each residue over time (Fig. 11A and B).
Fig. 11
A Information on the protein CYP19A1 shows that it has 464 total residues in chain A, 7485 total atoms, 3700 heavy atoms, and a charge of + 7. B Total residues in chain A of the protein CYP17A1 are 452, and there are 7399 total atoms, 3658 of which are heavy atoms with a charge of + 6
留言 (0)