
記住我


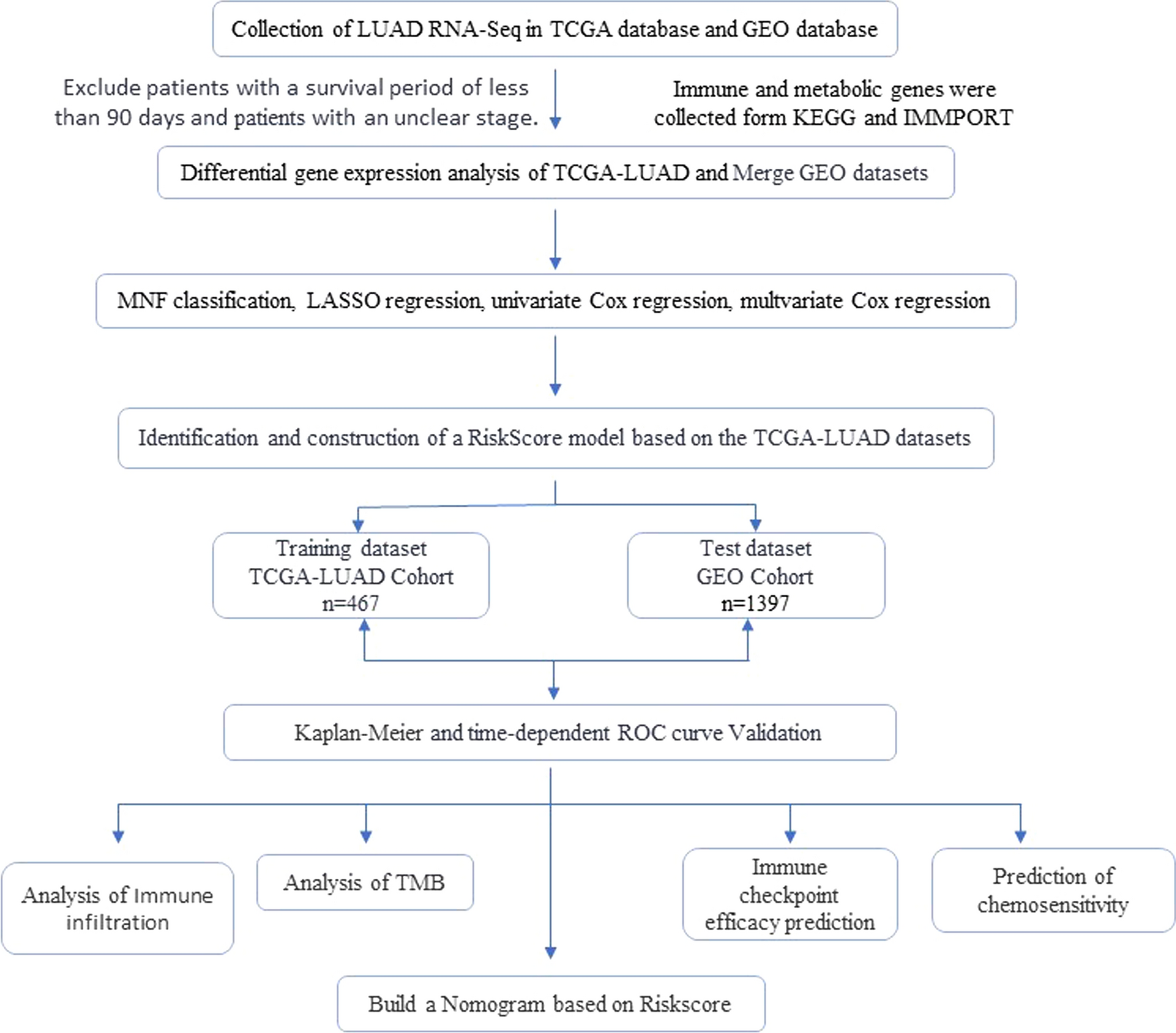


The overview of the process used in this study was shown in Fig. 1. After extraction from KEGG and MMPORT databases, a total of 947 genes related to metabolic pathways and 483 genes related to immunity were obtained. In the TCGA LUAD cohort (n = 502), 343 pairs of gene expression differences were found in contrast to adjacent normal tissues. The GEO-LUAD cohort was merged using the combat package and principal component analysis (PCA) was applied to evaluate the merging effect (Fig. 2). Through GEO cohort validation, 272 genes were identified that were expressed in both GEO and TCGA cohorts (Fig. 3A). Using these 272 pairs of genes for Non-negative matrix factorization (NMF) algorithm, the TCGA cohort was divided into two subtypes (Fig. 3B). According to the results of survival analysis, they were named high-risk group and low-risk group, and it was found that there were statistically significant differences in disease-free survival and survival between these two types (Fig. 3C).
Fig. 1 Fig. 2
Fig. 2
Principal Component Analysis (PCA) dimensionality reduction before merging GEO-LUAD Cohorts (A). Principal Component Analysis (PCA) dimensionality reduction before merging GEO-LUAD Cohorts (B)
Fig. 3
Construction of a NMF subtype based on Immune and Metabolic Related Genes in the TCGA-LUAD cohort. A Identification of DEGs between tumor and normal. B NMF consensus clustering for k = 2. C Kaplan–Meier analysis of overall survival (OS) for Cluster C1 and C2. D Kaplan–Meier analysis of disease-free survival (DFS) for Cluster C1 and C2. E ESTIMATE for Cluster C1 and C2
We used the ESTIMATE algorithm to estimate the population abundance of immune cells and stromal cell populations infiltrating the tissues in both subtypes. The ESTIMATE evaluation results showed that C1 subtype had a higher score compared to C2 subtype, indicating a more significant level of immune cell infiltration in C1 subtype (Fig. 3E).
3.2 Establishment and validation of prognostic modelsIn order to further explore the value of differential genes, The TCGA-LUAD cohort was randomly divided into a training group and a testing group in a 7:3 ratio. we combined univariate Cox regression, LASSO regression (Fig. 4A, B) and multivariate Cox regression (Fig. 4C), used to select 7 genes to construct an immune prognostic risk model. Risk scores were obtained by combining expression levels and regression coefficients (Riskscore = CAT exp × -0.166751936312374 + CCL20 exp × 0.0563504470190809 + GPI exp × 0.133661988590366 + INSL4 exp * 0.126270665159998 + NT5Eexp * 0.105058801123867 + GSTA3exp × -0.064669483229661) + GNPNAT1 exp × 0.136377362257813). The training set and test set patients were divided into low-risk group and high-risk group based on median risk score, and the robustness of the prognostic risk model was evaluated. In both TCGA cohort (Fig. 5A, B, C) and GEO cohort (Figure D), patients in the high-risk group had shorter survival time than those in the low-risk group ROC curves showed that Riskscore had the largest area under AUC curve among all clinical features (Fig. 5E). In TCGA cohort, the areas under AUC curve at 1 year, 3 years, and 5 years were: 0.752, 0.755, and 0.654 respectively (Fig. 5F).
Fig. 4
Screening for prognostic genes in LUAD using the TCGA-LUAD cohort.Lasso regression (A, B), multivariate Cox regression analyses (C)
Fig. 5
We were performed to screen candidate genes. Kaplan–Meier curves demonstrated the overall survival of LUAD patients in the Riskscore-high and -low groups of TCGA-LUAD train cohort (A), TCGA-LUAD test cohort (B), TCGA-LUAD cohort (C) and e GEO-LUAD cohort (D). The ROC curves to validate the accuracy of the risk model in predicting the clinical outcomes of patients with LUAD (E, F). All of statistical tests were performed using Cox regression analysis
According to the Riskscore, calculate the scores of each sample and then divide the GEO cohort and TCGA cohort into two groups based on the median risk score (Fig. 6A, B). Compare the gene expression patterns in different risk groups between the two cohort. The results show that regardless of whether it is in different risk groups in TCGA or GEO cohort similar abnormal gene expressions are observed in both (Fig. 6C, D). Additionally, there is a higher distribution of death cases in the high-risk group (Fig. 6E, F). In addition, after dividing the TCGA cohort into two groups based on patient staging (I–II stage and III–IV stage), we found that the high-risk group exhibited shorter survival (Fig. 6G, H). After further evaluation, we conducted a thorough analysis of the clinical characteristics of two patient groups. In the TCGA-LUAD cohort, significant differences were revealed in gender grading, stage classification, T staging, and N staging between the high-risk and low-risk subgroups(Fig. 6I).
Fig. 6
Prognostic value of Riskscore. According to the median risk score, TCGA-LUAD cohort was divided into high-risk group and low-risk group. A GEO-LUAD cohort was divided into high-risk group and low-risk group. B Gene expression profiles associated with high and low-risk groups in the TCGA-LUAD cohort prognostic model (C). Gene expression profiles associated with high and low-risk groups in the GEO-LUAD cohort prognostic model (D). The scatter plots are for the TCGA-LUAD cohort and the GEO-LUAD cohort, respectively. E, F Disparities in survival rates persist among high and low-risk subgroups within both the early-stage (G) and Advanced stage (H) cohorts. Differences in clinical characteristics between the high-risk and low-risk groups (I)
3.3 Construction of a NomogramAfter univariate Cox regression and Multivariate Cox analysis, we found that the risk score is an independent risk factor and used it to construct a column chart for prediction (Fig. 7A, B). In the column chart, scores for various factors such as age, stage, and risk score were calculated, and the total score can be used as a predictive tool (Fig. 7C). At the same time, a calibration curve was plotted to measure the consistency between actual observed prognosis values and predicted values from the column chart (Fig. 7D). The performance of the column chart was evaluated using ROC curves with AUC values of 0.715 at 1-year survival period, 0.735 at 3-year survival period, and 0.739 at 5-year survival period (Fig. 7E).
Fig. 7
Diagnostic efficiency of the Nomogram model. A and B Forest plots of the A univariate and B multivariate Cox regression analyses in TCGA LUAD cohorts. A nomogram model constructed based on the TCGA-LUAD cohort was used to assess the 1-, 3-, and 5-year overall survival of LUAD patients. C The calibration plots for the internal validation of the nomogram predicting 1-, 3- and 5-year D. Time-dependent ROC curve demonstrated the ability of the model to predict overall survival at 1-, 3- and 5-year in LUAD patients (E)
3.4 Clinical differences between high-risk and low-risk groupsAfter analysis, it was found that there is a correlation between the genes used to construct the prognostic model and the risk scores (Fig. 8A), and there are also differences in gene expression between high-risk and low-risk groups (Fig. 8B–H). However, when dividing the high and low expression groups based on the median expression of each gene, we observed survival differences between the high and low expression groups of CAT\CCL20\GPI\GNPNAT1 in TCGA cohort (Fig. 9A–D).
Fig. 8
Model of gene expression and Riskscore relationships in the TCGA-LUAD cohort. A Differential expression of prognostic genes between high and low risk groups in TCGA-LUAD cohort (B–H)
Fig. 9
CAT\CCL20\GPI\GNPNAT1 expression and its relationship with overall survival in the TCGA-LUAD cohort (A–D)
The analysis of the correlation between Riskscore and tumor mutation showed that risk score was positively correlated with tumor (Fig. 10A), and there were differences in TMB between high and low risk groups (Fig. 10B). Further investigation into the relationship between TMB and survival time found that the high mutation group had a shorter survival time (Fig. 10C). According to the division of risk groups, the TCGA cohort was divided into four groups: high-risk high-TMB group, high-risk low-TMB group, low-risk high-TMB group, and low-risk low-TMB group. The results showed that the low-risk high-TMB group had the longest survival time, followed by the low-risk low-TMB group and the high-risk high-TMB group (Fig. 10D). In terms of gene mutations, the analysis of the top 10 genes TP53, TTN, MUC16, CSMD3, RYR2, LRP1B, ZFHX4, USH2A, KRAS and IRP2 showed that the mutation rates of each gene in the high-risk group were significantly higher than those in the low-risk group (Fig. 10E, F).
Fig. 10
The tumor mutation burden characteristics in low- and high-risk group. Correlation analysis between Riskscore and TMB. (A) The difference in TMB between two groups. B Survival difference between high- and low-TMB. C Survival analysis of TMB along with Riskscore. D Mutational landscape in the high-risk group. E The difference in TMB between two groups. F TMB: Tumor mutational burden
To further investigate immune infiltration, we employed the MCPcounter method. We calculated the high- and low-risk groups, risk scores, and the population abundance of stromal cell populations. The results showed a correlation between B-cell lineage, T-cell, myeloid cell, endothelial cell, and fibroblast with Riskscore (Fig. 11A). In the high- and low-risk groups, the abundance of myeloid cells, endothelial cells, fibroblasts, and neutrophils varied (Fig. 11B–E).
Fig. 11
Immuno-infiltration analysis and Riskscore relationships in the TCGA-LUAD cohort (A). The difference in myeloid cells, endothelial cells, fibroblasts, and neutrophils between low- and high-risk group in TCGA-LUAD (B-E)
3.5 Potential drug sensitivity analysisWe used TIDE and TICA models to predict clinical differences between high-risk and low-risk groups, and used IMvigor210 as an external validation set. According to TIDE scores, high-risk groups showed higher TIDE score and Worse treatment response than low-risk groups (Fig. 12A, B). According to TICA scores, there was no statistically significant difference in AIPS scores in PD1 positive groups, but in PD1 negative groups, regardless of CALT4 expression, low-risk groups had higher scores (Fig. 12C). External validation on the IMvigor210 cohort showed that patients classified as high-risk groups had shorter survival and (Fig. 12D). The efficacy of immunotherapy was also assessed. The high-risk group demonstrated a higher likelihood of disease progression (PD) and disease stability (SD), whereas the low-risk group exhibited a greater probability of achieving complete remission (CR) and partial remission (PR) (Fig. 12E).
Fig. 12
Immunotherapy response of low- and high-risk TCGA-LUAD cohort. A, B Difference of the TIDE . C TCIA score of the low- and high-risk group in the of CTLA4- PD1-, CTLA4- PD1 + , CTLA + PD1−, and CTLA + PD1 + subgroups. D Kaplan–Meier curves for Prognostic Model in the IMvigor210. E Differences in Riskscore between patients with CR/PR and individuals with SD/PD after treatment with ICIs in the IMvigor210
The OncoPredict was used to conduct drug sensitivity analysis on high- and low-risk groups, specifically for common chemotherapy regimens of lung adenocarcinoma. The results showed that there were varying degrees of response differences among commonly used chemotherapy drugs such as paclitaxel (Fig. 13A), docetaxel (Fig. 13B), cisplatin (Fig. 13C), gemcitabine (Fig. 13D), and vinorelbine (Fig. 13E) in the high- and low-risk groups. These drugs are commonly used chemotherapy drugs for lung adenocarcinoma and showed higher scores in the low-risk group.
Fig. 13
Prediction of chemotherapeutic drugs in high-risk and low-risk groups. A–E The boxplot shows drugs with different IC50 values between the high and low risk groups
留言 (0)