Hepatocellular carcinoma (HCC) ranks the sixth most common form of cancer worldwide, with about 870,000 cases reported in 2022 [1]. Hepatitis B virus (HBV), poor diet and inactivity, fungal toxins, non-alcoholic fatty liver disease (NAFLD), alcohol consumption and infection with the hepatitis C virus are major pathogenic factors for HCC [2,3,4,5]. Due to the unobvious early symptoms, most HCC patients are diagnosed at advanced stage. Surgical resection is the first choice for most patients, while postoperative recurrence and metastasis rates are particularly high [6, 7]. Despite the great advances achieved in chemotherapy, targeted therapy and immunotherapy, patients with HCC generally have a poor prognosis [8, 9]. Therefore, it’s urgent to explore the underlying molecular pathogenesis of HCC and to develop effective intervention strategies.
MicroRNAs (miRNAs) are single-stranded small RNA molecules about 21–23 bases in size, which are generated by the dicer enzyme processing of single-stranded RNA precursors about 70–90 bases in size with hairpin structure [10]. Lin-4 and let-7 were the first identified miRNAs, which were found in nematode worms [11]. Subsequently, several research groups identified hundreds of miRNAs in a variety of biological species, including humans, fruit flies, plants and so on [12, 13]. MiRNAs have high biological genome coding and degrades mRNA or blocks its translation by pairing with target gene mRNA base to guide silencing complex (RISC). Increasing evidence found that miRNAs play a variety of roles in the regulation of cell growth and development. In addition, numerous studies showed that miRNAs participated in the regulation of tumorigenesis, progression and drug resistance of human malignant tumors [14,15,16,17]. miR-3191 is a newly discovered miRNA, and its function and mechanism of action in biological processes and diseases are not completely understood [18]. It was reported that miR-3191 promotes migration and invasion by downregulating TGFBR2 in colorectal cancer [19]. Nonetheless, the role of miR‑3191 and its underlying mechanism in the progression of HCC remain poorly understood.
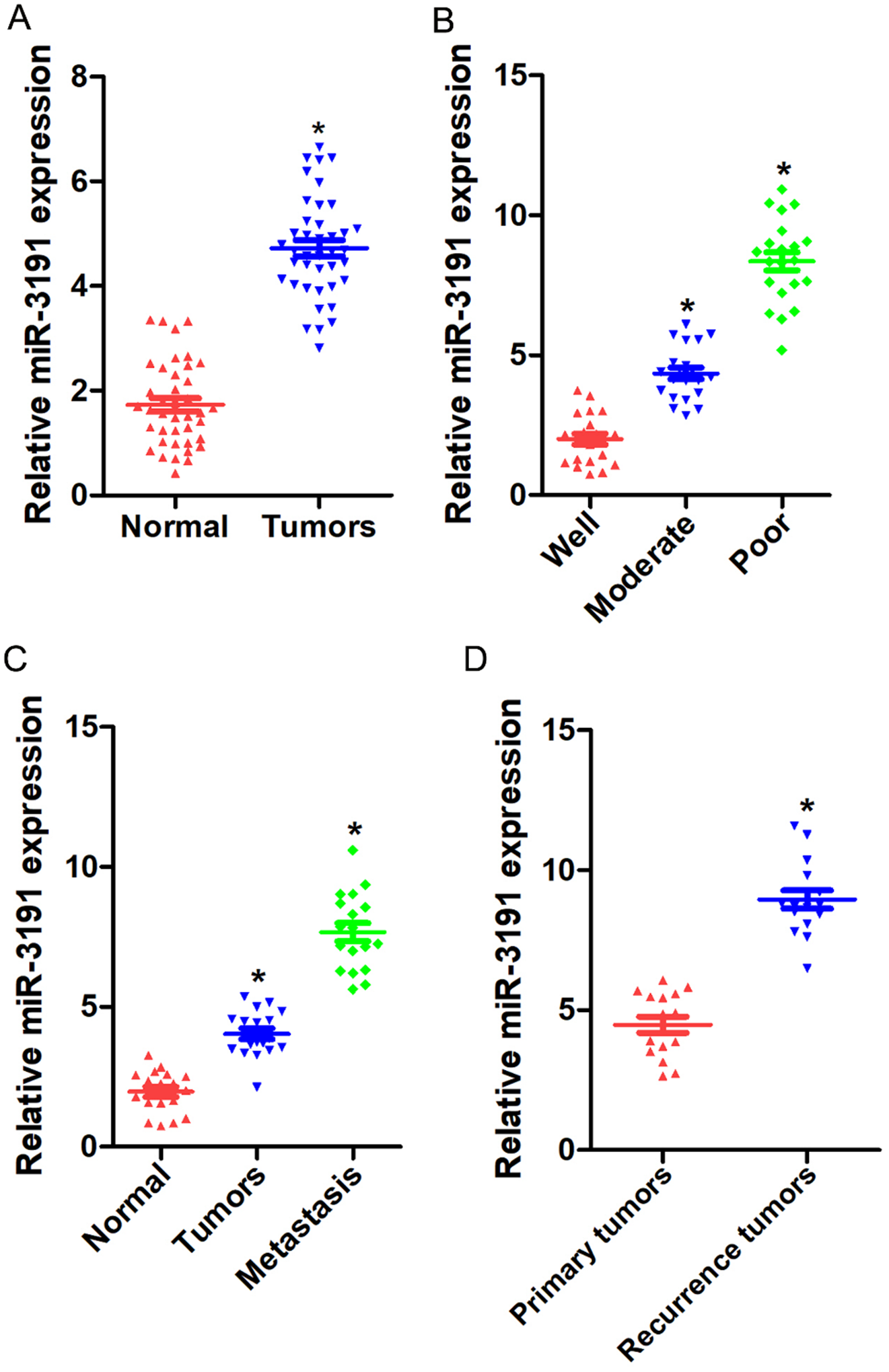
In this study, we for first find that miR-3191 is highly expressed in HCC tissues and predicts poor prognosis of HCC patients. Next, by using loss-of-function analysis and gain-of-function analysis in HCC cells, we demonstrate that miR-3191 promotes HCC cells proliferation and metastasis. Further mechanism study reveals that PAK6 is a direct target of miR-3191 in HCC cells. Clinical investigation also confirms the correlation between miR-3191 and PAK6, and demonstrates the value of combining miR-3191 and PAK6 to improve the prognostic accuracy for HCC patients. Altogether, we discover that miR-3191 promotes the proliferation and metastasis of HCC cells via targeting PAK6.
Materials and methodsPatients and samples
HCC samples were collected from patients who underwent the resection of their primary HCC in Eastern Hepatobiliary Surgery Hospital (EHBH, Shanghai, China). Cohort 1 patients (n = 180, from 2009 to 2013) and cohort 2 patients (n = 113, from 2011 to 2014) were collected to investigate the clinical significance of miR-3191 in HCC. Detailed clinicopathological features are described in supplementary Tables 1&2. Overall survival (OS) was defined as the interval between the dates of surgery and death. The recurrence was defined as the interval between the dates of surgery and recurrence; if recurrence was not diagnosed, then patients were censored on the date of death or on the last follow-up. Forty human HCC tissues and normal tissues, Eighteen paired peri-tumor normal tissue, primary HCC and metastatic foci, Sixteen paired primary HCC and recurrent HCC tissues, and Sixty-three differentially differentiated human HCC tissues were collected to detect miR-3191 expression. The patients’ informed consent was also obtained, and all procedures were approved by the ethical committee of EHBH.
Cell lines and cell culture
HCC cell lines Hep3B and Huh7 were provided by Erasmus University (Rotterdam, Netherlands). All human HCC cells were cultured in DMEM (Gibco®, New York, USA) with 10% FBS (Gibco®), glutamine and penicillin/streptomycin (Gibco®) at 37 °C in 5% CO2 incubator. Mycoplasma contamination was excluded via a PCR (D7232, Beyotime, Shanghai, China) based method. The cell lines were authenticated by short tandem repeat (STR) DNA profiling.
The lenti-vector expressing miR-3191 mimic, miR-3191 inhibitor and their control virus were purchased from Shanghai GenePharma (Shanghai, China). NC mimic: 5’-UUCUCCGAACGUGUCACGUTT-3’, miR-3191 mimic: 5’-UGGGGACGUAGCUGGCCAGACAG-3’, NC inhibitors: 5’-CAGUACUUUUGUGUAGUACAA-3’, miR‐3191 inhibitors: 5’-CUGUCUGGCCAGCUACGUCCCCA-3’. siPAK6 and its control siRNA were purchased from Shanghai GenePharma. Huh7 and Hep3B cells were infected with miR-3191 mimic, miR-3191 inhibitor and their control virus and the stable infectants were screened by puromycin as described previously [20].
Cell counting kit-8 (CCK-8) assay
CCK-8 assay was performed according to the manufacturer’s instructions to measure cell proliferation. Then, 3 × 103 HCC cells were plated and incubated in 96-well plates for 0, 24, 48, 72 and 96 h. They were placed in an incubator containing 5% CO2 at 37 °C. Next, 10 µL of CCK-8 reagent was added to each well for 1 h (Dojindo Molecular Technologies, Kumamoto, Japan). Finally, they were detected using a microplate reader (Molecular Devices, Kumamoto, Japan) at an absorbance of 450 nm.
Colony formation assay
For colony formation assay, HCC cells were cultured in 96-well plates with 1,000 cells/well at 37 °C for 7 days, and were subsequently stained with 0.1% crystal violet (C0121, Beyotime, Shanghai, China). Then, a light microscope (ThermoFisher Scientific, USA) was used to count the colonies.
5-ethynyl-2’-deoxyuridine (EdU) immunofluorescence staining
For cell EdU immunofluorescence staining, HCC cells were seeded into 96-well plates (3 × 103 cells) and performed using the EdU Kit (C0071S, Beyotime, Shanghai, China). The results were quantified with a Zeiss axiophot photomicroscope (Carl Zeiss) and Image-Pro plus 6.0 software.
Cell metastasis assays
For cell migration experiments, 2 × 105 HCC cells were seeded into the upper chamber of a polycarbonate transwell in serum-free DMEM medium. The lower chamber was added with DMEM medium containing 20% FBS as chemoattractant. The cells were incubating for 16 h and the chamber was fixed with 10% neutral formalin for more than 4 h. The cells were dyed with crystal violet. The cells were then counted under a microscope (Olympus) and the cell number is expressed as the average number of the cells in each field.
For cell invasion experiments, 2 × 105 HCC cells were seeded into the upper chamber of a polycarbonate transwell in serum-free DMEM medium. The lower chamber was added with DMEM medium containing 20% FBS as chemoattractant. The cells were incubating for 36 h and the chamber was fixed with 10% neutral formalin for more than 4 h. The cells were dyed with crystal violet. The cells were then counted under a microscope (Olympus) and the cell number is expressed as the average number of the cells in each field.
Animal models
BALB/c nude mice (male, 6 weeks old) were purchased from Chinese Academy of Sciences Slack Company (Shanghai, China). For xenograft formation assay, 2 × 106 Huh7 miR-3191 inhibitor cells and control cells were injected subcutaneously into nude mice (n = 6 each, randomized allocated). Nude mice were sacrificed six weeks post inoculation and tumors were collected and xenografts weight examined.
For lung metastatic model, a total of 2 × 106 Huh7 miR-3191 inhibitor cells and control cells were injected into the tail veins of nude mice (n = 6 each, randomized allocated). Mice were sacrificed at 2 months post injection. The lung tissues of each mouse were separated and subjected to H&E staining. And the lung metastatic foci were counted in a double-blind manner with the aid of a dissecting microscope. All animal experiments were approved by the Animal Care Committee of Community Health Service Center.
Real-time PCR
Real-time PCR were carried out as described in our previous paper [20]. Briefly, total RNA was extracted using an miRcute miRNA isolation kit (TIANGEN, Beijing, China) for miRNA and TRIzol (Life Technologies, USA) for mRNA in accordance with the manufacturer’s instructions. Then, miRNA and mRNA were polyadenylated using a poly-A polymerasebased First-Strand Synthesis kit (Takara, Kyoto, Japan). Subsequently, a PrimeScript RT Reagent kit (TaKaRa) were used for reverse transcription (RT) of the total mRNA. Finally, miR-3191 and complementary DNA (cDNA) in these samples were assayed using SYBR Green I (Applied Biosystems, USA). The primer sequences were: PAK6 primer sequences were forward: 5’ GCTCTCGGACTTCGGATTCT 3’, reverse: 5’ GGCATACAAAGACCTGGAGAT 3’. The β-actin was used as reference for relative expression calculation and its primer sequences were forward: 5’ GGCCCAGAATGCAGTTCGCCTT 3’, reverse: 5’ AATGGCACCCTGCTCACGCA 3’.
Western blotting assay
Protein extraction from HCC cells were finished by using the RIPA lysis buffer reagent (P0013B, Beyotime, Shanghai, China), and the protein quality was determined by BCA kit (P0009, Beyotime). Next, proteins were separated by SDS-PAGE, transferred onto the PVDF membranes (Millipore, USA), and incubated with primary antibodies against PAK6, N-cadherin, E-cadherin, and Vimentin, and subsequently with the secondary antibodies. Finally, an electrochemiluminescence (ECL) system (5200CE, Tanon, Shanghai, China) was used for protein bands visualization, which were analyzed by using the Image J software. The primary antibodies were E-cadherin (1:1000; 20874-1-AP, Proteintech), N-cadherin (1:1000; 22018-1-AP, Proteintech), PAK6 (1:1000; 13539-1-AP, Proteintech) and GAPDH (1:5000; #5174, Cell Signaling Technology).
Luciferase reporter assay
The 3’-untranslated region (3’-UTR) of wildtype (GUCCCCA) or mutant (ACAGUAC) PAK6 was inserted into the pmir-GLO Luciferase vector (Promega, Madison, WI, USA) for Luciferase reporter experiments. Then, the above Luciferase plasmids were transfected into miR-3191 mimic or inhibitor and their control HCC cells. Finally, cells were extracted with passive lysis buffer [25 mM Tris-phosphate (pH 7.8), 2 mM EDTA, 1% Triton X-100, and 10% glycerol]. The luciferase activity was measured with a microplate luminometer LB 96 V (Berthold GmbH & Co. KG, Bad Wildbad, Germany). The Renilla luciferase signal was normalized to the internal firefly luciferase transfection control.
RNA immunoprecipitation (RIP) assay
The RIP assay were used to verify the binding relationship between miR-3191 and PAK6. The RIP assay was performed using the Magna RIP™ RNA Binding Protein Immunoprecipitation Kit (Millipore, USA) according to the manufacturer’s instructions. Cells were lysed in RIP lysis buffer, and then were incubated with RIP buffer with a human anti-Ago2 antibody (Abcam, Cambridge, MA, USA). Each sample was incubated with proteinase K to digest protein. Purify RNA was obtained and then was analyzed using qRT-PCR assay.
Immunohistochemistry
The tissue samples were fixed with 10% neutral formaldehyde, embedded in paraffin, and sectioned for immunohistochemical (IHC) staining. In brief, after antigen retrieval, sections or tissue microarrays (TMAs) were blocked with bovine serum antigen albumin (BSA) and incubated with the indicated primary antibody and then secondary antibody. A diaminobenzidine (DAB) colorimetric reagent solution was used, followed by hematoxylin counterstaining. The slides were scanned, and representative images were captured. IHC scoring was based on the percentage of positively stained cells, and staining intensity was assessed by Image Scope software (Aperio Technologies, Inc.).
Statistical analysis
Each experiment was repeated at least three times. Statistical analysis was performed using SPSS V.18.0. The data are expressed as the mean ± SD. Two-tailed Student’s t test or the Mann-Whitney U test was used to compare two continuous variables, and the chi-square test was used to compare qualitative variables. The DFS and OS of distinct subgroups were compared by the Kaplan-Meier method and log-rank analysis. Pearson’s correlation analysis was performed to determine the correlation between two variables. Each dataset was analyzed separately. A p value less than 0.05 was considered statistically significant.



留言 (0)