
記住我
Idiopathic Pulmonary Fibrosis (IPF) is an idiopathic, progressive, and chronic illness that causes an accumulation of scarred tissue in the lungs, leading to difficulty breathing and disseminating oxygen into the bloodstream. Idiopathic pulmonary fibrosis is typically seen in people over the age of 50 (Lederer and Martinez, 2018). In the United States, the adjusted prevalence is estimated to be 2.4 cases per 10,000 people (Nalysnyk et al., 2012; Maher et al., 2021). The prognosis is poor, and most individuals only survive 2-3 years following diagnosis (Strongman et al., 2018). The majority of IPF cases are sporadic and occur with no prior family history of this disease. However, there are still instances of familial pulmonary fibrosis, which is believed to exhibit a pattern of autosomal dominant inheritance, wherein a single copy of the altered gene is sufficient to cause the illness. Even then, there are individuals who, despite inheriting the altered gene, do not go on to develop fibrosis. The reasons for this remain unclear. Occupational exposures can also increase the likelihood of developing idiopathic pulmonary fibrosis; farming, agricultural professions, and pesticide contact raise the risk of IPF development (Figure 1). Smoking and past exposure to metal or wood dust are also associated with higher rates of IPF (Paolocci et al., 2018; Park et al., 2021).

Figure 1. Pathogenesis of Idiopathic Pulmonary Fibrosis (IPF) and microbial persistence. The etiology of IPF includes a multitude of triggers to the homeostatic lung functioning. Occupational and chronic exposure to particulate matter, allergens, persistent microbial infections, age-associated inflammation and senescence, and genetic predisposition lead to repetitive alveolar injury and increase the risk of developing IPF. Alveolar epithelial cells (AEC) play a central role in maintaining the alveolar-vascular barrier and initiating repair after injury, while antigen-presenting cells (APC) mediate immune responses to microbial infections. Compared to healthy lungs with normal gas exchange and alveolar-vascular barrier integrity, persistent microbial infection and intracellular inhabitance may lead to abnormal alveolar repair with vascular leakage and uncontrolled inflammation. These aberrant alveolar/vascular functions further lead to mucus deposition and fibroblast activation to produce extracellular matrix (ECM) components, causing scarring of the lungs and poor gas exchange, thereby declining the overall lung function.
Overall, 20% to 50% of all ILDs exhibit clinical manifestations of IPF, presenting unique histological phenotypes, including usual interstitial pneumonia, areas of honeycombing, and fibroblastic foci. To date, two antifibrotic therapies that have been FDA-approved and are available to treat IPF include Pirfenidone and Nintedanib. Studies have shown that Pirfenidone and Nintedanib effectively inhibit fibroblast proliferation and activity and the deposition of extracellular matrix proteins in the lungs, both critical processes in developing lung fibrosis (Conte et al., 2014; Wollin et al., 2015). To that end, these drugs indeed work to slow the progression of the disease. However, they do not reverse the fibrotic damage that has already occurred, nor do they completely halt disease progression. Clinical studies and real-world data suggest that both Pirfenidone and Nintedanib offer modest improvements in lung function and survival but fall short of providing a cure. Their efficacy is limited to reducing the rate of decline in lung function. Moreover, both drugs are associated with significant side effects, including gastrointestinal disturbances, liver dysfunction, nausea, and fatigue, which can impact patients’ quality of life and lead to treatment discontinuation (Finnerty et al., 2021; Kou et al., 2024). This underscores the need for more effective therapies that not only slow disease progression but also reverse existing fibrosis and improve overall outcomes for patients.
Furthermore, abundant research has studied associations between IPF and viruses, such as human metapneumovirus, influenza virus, and coronaviruses. Idiopathic pulmonary fibrosis patients are coinfected with viral and bacterial infections show significantly diminished lung function and increased risk of mortality (Moghoofei et al., 2022). However, the contributions of persistent bacterial infections in IPF are understudied. The main body of this review seeks to elucidate connections between these infections and specific mechanisms utilized by their causative pathogens, which may accelerate or complement fibrotic processes in IPF. It examines the role of Gram-positive and Gram-negative bacteria infections and highlights promising avenues for future investigation into possible treatments and diagnostic methods.
Here, we discuss how viral, fungal, and bacterial infections contribute to the pathogenesis of IPF, review the literature, and identify areas where further investigation is necessary, with an emphasis on the commensal Gram-negative bacterium Haemophilus influenzae. It is among the most common causes of upper and lower respiratory tract infections in adults and children (Ieven et al., 2018; Lv et al., 2024). The non-typeable Haemophilus influenzae (NTHi) exacerbates the pathogenesis of chronic obstructive pulmonary disease (COPD), airway inflammation, and end-stage lung diseases (Su et al., 2018; Saliu et al., 2021). For example, overactivity of the mucin-producing gene MUC5B is associated with goblet cell dysfunction in COPD and IPF (Molyneaux et al., 2014; Hanmandlu et al., 2022; Huang et al., 2022). Chronic middle ear infection with NTHi is also associated with dysregulation of MUC5B mucins (Val et al., 2015). However, the mechanisms of NTHi intracellular pathogenicity are underexplored in the progression of lung diseases. As for COPD, we suggest here that persistent intracellular NTHi infection should be investigated in the context of progressive IPF development.
Altogether, this review addresses a critical gap in the literature, as no recent or comprehensive analyses have thoroughly examined this specific topic. Unlike a systematic review, which emphasizes clinical research and methodology, this work offers an in-depth perspective on mechanistic insights and the underlying processes that could intrigue future therapeutic strategies.
2 Viral Infections in IPFNumerous studies have shown that viral infections may play a key role in the pathogenesis of IPF (Sheng et al., 2020). Most of these studies have looked explicitly at Epstein-Barr virus (EBV) and other herpesviruses in relation to IPF. In a serological study, Manika et al. reported that 60% of patients with IPF had anti-EBV immunoglobulin A (IgA) (P=0.024), compared to only 22% of control patients (Manika et al., 2007). Additionally, the group found EBV DNA via polymerase chain reaction (PCR) in the bronchoalveolar lavage (BAL) fluid of 3 of 17 patients with IPF. A study by Kelly et al. investigated the presence of WZhet, a rearranged genetic frame of EBV associated with productive EBV replication, in both IPF patients and controls (Kelly et al., 2002). Despite 75-85% of patients across all groups testing positive for EBV DNA by buffy coat analysis, 0% and 4% of the two control groups were positive for WZhet, whereas 59% of IPF patients had WZhet present in peripheral blood samples. Since 61% of EBV-positive lung biopsies in IPF patients also demonstrated WZhet presence, evidence indicates a correlation between WZhet expression in lung tissue and peripheral blood. Despite broad evidence for the role of immunosuppressive therapy in reactivating herpes viruses, this study noted no relationship between prior immunosuppressive treatments and WZhet expression. These data further confirm the association between active EBV infection and IPF, suggesting a potential marker in the peripheral blood for tracking EBV in this disease. Numerous studies have reported the presence of viral DNA in IPF patients, such as the detection of EBV by PCR and immunohistochemistry in 11/27 of patients with IPF but not in controls (Stewart et al., 1999). Furthermore, Herpesvirus saimiri DNA, a virus naturally found in squirrel monkeys, was also detected in the regenerating epithelial cells of 21/21 IPF biopsy samples examined (Folcik et al., 2014). The latter case is of particular interest, given that H. saimiri’s low infection rate in humans (around 7%) aligns better with its potential role as an etiological factor in a rare disease like sporadic IPF (Moore and Moore, 2015). Murine gammaherpesvirus-68 (MHV-68) also shares significant homology with H. saimiri, and it is a widely used strain for preclinical studies on the effects of herpesvirus on lung fibrosis (Bortz et al., 2003; Folcik et al., 2014). Altogether, these studies point to herpesviruses as potentially significant in the pathogenesis of IPF.
Among the first studies to propose a mechanism for the viral pathogenesis of IPF is one that investigates the role of malfunctioning type II alveolar epithelial cells (AECs) in IPF patients, showing the presence of herpes virus protein (Lawson et al., 2008). Specifically, the group investigated the role of surfactant protein C (SFTPC) mutations in IPF, examining how mutant SFTPC expression may induce endoplasmic reticulum (ER) stress and the unfolded protein response (UPR) in AECs. They found herpesvirus proteins in AECs from 15/23 IPF patients, which colocalized with UPR marker XBP-1 (X-box binding protein-1), indicating a potential link between herpesvirus infection, ER stress, and IPF progression (Lawson et al., 2008). This suggests that ER stress and UPR activation in the alveolar epithelium could contribute to the development and worsening of IPF, possibly through chronic herpesvirus infection or altered surfactant protein processing. Systemic sclerosis-associated IPF patients also showed qPCR positivity results for the presence of EBV latent membrane protein-1 (LMP1) that could be associated with more rapid disease progression in IPF. In a one-year follow-up study, it was reported that a large number of LMP1-positive patients with IPF died from respiratory failure as compared to LMP1-negative patients, suggesting that EBV LMP1 could play a role in the progression of IPF (Tsukamoto et al., 2000). A case study reinforced these findings, indicating that latent LMP1-positivity correlated with poor prognosis (Marzouk et al., 2005). Idiopathic pulmonary fibrosis patients also demonstrated EBV and p53 expression via immunohistochemistry, compared to the absence of expression in the control group (P = 0.05). This suggested that a functional relationship between EBV and p53 may exist in patients with IPF, as p53 activity is central in regulating the cell cycle and apoptotic cell death (Lok et al., 2001). Intriguingly, administering the antiviral medication Ganciclovir to advanced IPF patients with signs of previous EBV showed promising results, weakening the progression of IPF (Egan et al., 2011).
Preclinical studies have helped us gain deeper insights into the mechanisms by which a gamma herpesvirus (γHV) infection can influence the development of lung fibrosis. The first example of herpesvirus-inducing fibrosis in a natural host came from a study by Williams et al., who showed that infecting horses with an equine γHV led to the development of fibrosis (Williams et al., 2013). In murine models, prior infection with MHV-68, even in its latent state, can amplify lung fibrosis triggered by subsequent fibrotic insults with bleomycin or fluorescein isothiocyanate (Vannella et al., 2010). This augmentation of fibrosis seems unrelated to active viral replication. Moreover, latent viral infection in mouse lungs correlates with heightened production of chemokines attracting fibrocytes and inflammatory cells and increased release of cysteinyl leukotrienes, tumor necrosis factor α, and profibrotic transforming growth factor β1 (TGF-β1), suggesting potential profibrotic mechanisms involved in viral influence on pulmonary fibrosis (Vannella et al., 2010; Stoolman et al., 2011). While these studies have demonstrated that prior infections may increase susceptibility to subsequent fibrotic injury by altering lung epithelial cells during latency and prompting their secretion of profibrotic factors, viral infection can also exacerbate fibrotic disease when superimposed on pre-established fibrosis (McMillan et al., 2008). In such cases, active lytic replication correlates with increased TGF-β1 signaling and epithelial cell apoptosis, ultimately resulting in increased collagen deposition within the lung. Notably, not all viral infections cause the same degree of fibrosis in the lung. For example, MHV-68, but not influenza A (H1N1), was able to exacerbate lung fibrosis in mice, indicating there may be some specificity involved (Ashley et al., 2014).
Several studies have investigated the role of other viruses in IPF, although most have found little to no association. For instance, despite adenovirus’ capacity to stimulate the secretion of TGF-β1 from epithelial cells and induce epithelial-mesenchymal transition (EMT) via its E1A protein, adenovirus activity has ultimately shown limited correlation with IPF (Hayashi and Hogg, 2007). Similarly, the prevalence of adenovirus, enterovirus, or bocavirus DNA in lung biopsy samples, nasopharyngeal swabs, and bronchioalveolar lavage (BAL) fluids did not correlate significantly with the induction of profibrotic transformation in IPF patients compared to controls (Kuwano et al., 1997; Moradi et al., 2017).
The hepatitis C virus (HCV), which is known to trigger liver cirrhosis with chronic infection, has also been explored in the studies of lung fibrosis. A positive association between HCV and IPF was observed, with increased HCV antibody levels detected in IPF patients compared to controls (Ueda et al., 1992; Zidan et al., 2015), and increased incidence of IPF in patients previously infected with HCV compared to those infected with hepatitis B virus (Arase et al., 2008). Elevated anti-HCV antibody levels were also shown to be consistent across other lung diseases, suggesting it may not be exclusive to IPF (Irving et al., 1993; Meliconi et al., 1996).
In a 2001 study testing the sera of 33 IPF patients, the recently identified Torque teno virus (TTV) was present in 36.4% of patients. Additionally, the TTV-positive group exhibited a significantly worse 3-year survival rate (58.3%) than the TTV-negative group (95.2%) (Bando et al., 2001). Another study found that TTV was the most common virus in patients with acute exacerbation of IPF and was also present in patients with acute lung injury (Huie et al., 2010; Wootton et al., 2011). However, another study measuring TTV DNA titers in patients with acute exacerbation of IPF suggested that an association between TTV and the onset of acute exacerbation of IPF was unlikely (Bando et al., 2015).
Furthermore, the proposed mechanism of SARS-CoV-2-induced pulmonary fibrosis has striking similarities to fibrotic processes observed in IPF. The virus damages alveolar epithelial cells type 2 (AEC2), inciting macrophage activation and causing additional injury to the alveolar basement membrane. Macrophage response and AEC2 injury precipitate the release of inflammatory regulators such as IL-6, TNF-α, and TGF-β1. However, it is TGF-β1 that primarily acts to encourage fibroblast proliferation and differentiation into myofibroblasts (Alrajhi, 2023; Duong-Quy et al., 2023). Patients with IPF also have increased pulmonary levels of TGF-β1, and TGF-β1-mediated stimulation of the epithelial-to-mesenchymal transition is widely believed to contribute to progressive fibrosis (Wolters et al., 2014). Current studies center on the effectiveness of antifibrotic agents classically used for IPF in treating post-COVID-19 pulmonary fibrosis (Patrucco et al., 2023).
Nonetheless, host antiviral mechanisms such as the mitochondrial antiviral signaling protein (MAVS) may also contribute to the pathogenesis of IPF. In bleomycin-induced pulmonary fibrosis, mice showed increased MAVS response to damage-associated molecular patterns (DAMPs) generated by bleomycin injury. By mimicking the BH3 components, MAVS- downstream targets B-cell lymphoma-2 complex (Bcl-2), and Bcl-xl pro-apoptotic proteins attenuated MAVS-mediated fibrotic pathology (Kim et al., 2021). It is noteworthy that Bcl-2 and Bcl-xl complex also mediate anti-apoptotic and anti-proliferative effects as a unique dual-cell cycle response that may be responsible for the BH3 mimetic-mediated attenuation of fibrosis (Janumyan et al., 2003). Strikingly, the SARS-CoV2 nucleocapsid protein activates the Bcl-2 family protein, myeloid-cell leukemia-1 protein (MCL-1), to inhibit apoptosis, enhancing viral propagation and infectivity (Pan et al., 2023). In contrast, SARS-CoV2 manipulates MAVS signaling by several non-specific proteins from the open reading frames ORF3a, ORF9, and ORF10, thereby leading to increased lung tissue injury and non-resolvable COVID-19 interstitial lung disease (Wu et al., 2021; Wang et al., 2022). Another antiviral response protein, the engulfment and motility (ELMO) domain containing-2 protein (ELMOD-2), is also genetically implicated in IPF pathology. The antiviral mechanisms of ELMOD-3 were elucidated using the overexpression and knockdown strategies in A549 alveolar epithelial cells and demonstrated to be mediated by intracellular TLR3 signaling during influenza viral infections (Hodgson et al., 2006; Pulkkinen et al., 2010). Interestingly, SARS-COV2 infection seems to downregulate the expression of ELMOD2 and, therefore, ELMOD-2-mediated antiviral response (Radzikowska et al., 2023).
While there certainly are similarities in fibrotic pathways between IPF and SARS-CoV-2-related pulmonary fibrosis, it is critical to recognize their distinct clinical trajectories. SARS-CoV-2 infection is known to induce fibrotic changes, often evident on CT scans, particularly in patients with severe respiratory illness. However, longitudinal studies have demonstrated that these fibrotic changes largely resolve over time in the majority of patients, especially those with mild-to-moderate disease (Ahamed et al., 2024; Cortes-Telles and Zavorsky, 2024; Pinto et al., 2024). Unlike IPF, where fibrosis progresses inexorably and irreversibly, post-COVID-19 fibrosis appears transient and more reflective of reparative mechanisms following acute lung injury.
The differences in outcomes are underpinned by diverging pathophysiological processes. Idiopathic pulmonary fibrosis is characterized by chronic, progressive scarring driven by genetic predispositions, environmental exposures, and dysregulated fibroblast activity. In contrast, SARS-CoV-2-related fibrosis is primarily an acute inflammatory response to alveolar epithelial damage, often mediated by cytokine storms and endothelial injury. Shared pathways, such as TGF-β1 signaling and epithelial-to-mesenchymal transition, are activated in both conditions; however, their duration and impact differ significantly. For example, TGF-β1 signaling in IPF is sustained, contributing to irreversible extracellular matrix deposition (Kim et al., 2018), whereas in SARS-CoV-2, it may subside with the resolution of inflammation (Alfaro et al., 2024). Still, SARS-CoV-2 fibrosis provides an opportunity to explore mechanisms involved in acute fibrotic processes, which may share some overlap with pathways observed in the chronic progression of IPF. Although the outcomes of fibrosis in these two conditions often differ, studying these shared mechanisms may inform the development of therapeutic strategies, such as TGF-β1-targeted therapies in the treatment of COVID-19 and IPF (Budi et al., 2021; P et al., 2021).
3 Fungal Infections in IPFLittle is presently understood about the role of fungal colonization and infection in IPF and interstitial lung disease development (Clarke et al., 2018). Implicated species requiring further investigation include Pneumocystis jirovecii, Aspergillus fumigatus, and Candida albicans. A comparison of fungal microbiomes between patients with IPF and controls identified Pneumocystis jirovecii as the predominant fungal species in two stable IPF subjects and six patients with acute exacerbations of IPF. Importantly, P. jirovecii was not detected in any of the 40 control patients tested, highlighting its specific association with IPF (Molyneaux et al., 2016). In addition, a study examining 18 IPF patients discovered P. jirovecii colonization in 27.8% of the patient population tested (Vidal et al., 2006). While the sample sizes in these studies are relatively small, the results provide an intriguing lead point for future investigation.
Aspergillus fumigatus is a known instigator of complications in patients with IPF; one investigation of its association with interstitial pneumonia found that 9 out of the 15 patients in the study with diagnosed pulmonary aspergillosis had IPF. The remaining 6 had non-IPF interstitial pneumonia (Kurosaki et al., 2014). The role of A. fumigatus is also evident in triggering an acute exacerbation of IPF in a patient with no other diagnosed medical conditions who actively took an antifibrotic agent that had previously stabilized pulmonary deterioration (Suzuki et al., 2018).
Importantly, Roudbary et al. showed that C. albicans was the most prevalent fungal species detected in BAL samples collected from patients with IPF. These results are intriguing because none of the individuals in this patient cohort showed any clinical indications of fungal infection for the duration of the study (Roudbary M et al., 2019). While any direct mechanism of C. albicans to IPF development in humans beyond the promotion of scarring via repeated lung insults has yet to be thoroughly investigated, an experiment looking at bleomycin-induced pulmonary fibrosis in mice found that intestinal overgrowth of C. albicans correlated with exacerbated presentations of the illness. The proposed mechanism for this aggravation is an endothelial-to-mesenchymal transition mediated by IL-17A (Fukuda et al., 2018; Yamada et al., 2023).
Finally, recent evidence has also indicated that inoculation with the fungus Paracoccidioides brasiliensis can induce experimental pulmonary fibrosis in mice (Franco et al., 1998; Gonzalez et al., 2008). The potential benefits of antifungal therapy in treating IPF are even less thoroughly investigated than the contribution of the mycobiome to disease pathogenesis. Still, a combination of anti-fungal itraconazole and the anti-vascular disease drug pentoxifylline therapy significantly reduced inflammation and pulmonary fibrosis in these mice (Naranjo et al., 2011).
4 Bacterial infections in IPF4.1 Lung microbiome in pulmonary fibrosisIn recent years, altered lung microbiomes have been associated with IPF (Hewitt and Molyneaux, 2017; Puiu et al., 2024). Lung dysbiosis contributes to pulmonary inflammation by elevation of alveolar profibrotic cytokines (O’Dwyer et al., 2019) and modulation of the host immune response (Fabbrizzi et al., 2021). Pre-clinical studies of bleomycin-induced fibrosis in germ-free mice revealed that the lack of a microbiome attenuated mortality related to fibrotic injury. These results suggest that modification of lung microbiota could serve as a new approach for treating IPF (O’Dwyer et al., 2019; Fabbrizzi et al., 2021).
A Correlating Outcomes with biochemical Markers to Estimate Time-progression (COMET) study in IPF examined 55 BAL fluid samples (Han et al., 2014). DNA analysis revealed a positive association with Staphylococcus and Streptococcus genera and progression of IPF. The precise identification of bacterial species was impossible; nevertheless, the study effectively conveyed that certain Staphylococcus and Streptococcus operational taxonomic units (OTUs) are linked to worse IPF outcomes. Another study compared the bacterial burden in the BAL contents of 65 IPF patients with that of 44 controls (Molyneaux and Maher, 2014; Molyneaux et al., 2014). Not only did they find double the burden of bacteria in the BAL of the IPF patients, but they also found a strong association between patients with higher bacterial burden detected in their BAL and a decline in lung function and death. Furthermore, the study identified that the OTUs Streptococcus alongside Gram-negatives, Haemophilus, Neisseria, and Veillonella spp. were linked to IPF.
A separate analysis of BAL samples from IPF patients sought to correlate the microbiome with host immune response signaling pathways. Inflammation through fibroblast function and leukocyte phenotypes was assessed, revealing that some IPF patients exhibited changes in microbial diversity and that the lung microbiome is particularly associated with genes involved in the immune response, such as inflammation and tissue remodeling (Huang et al., 2017). Although causality was not established, the study suggests microbial influence on innate immunity and fibrosis. Other culture-independent studies have also shown an increased bacterial burden in the BAL of IPF patients (Spagnolo et al., 2019), particularly those experiencing acute exacerbation of the disease (Molyneaux et al., 2017; Invernizzi and Molyneaux, 2019). One study implicated the toll-like receptor 3 L412F polymorphism in dysregulating the lung microbiome and reducing the immune response to bacterial infection, leading to increased acute exacerbation associated-IPF death (McElroy et al., 2022).
DNA analysis reveals several distinct microbial signatures of IPF showed a characteristic abundance of Streptococcus, Pseudobutyrivibrio, and Anaerorhabdus. Microbial gene functionality related to ABC transporter systems (ATP synthase (ATP)-binding cassette transporters), biofilm formation, and two-component regulatory systems were more prevalent in the microbiome of IPF patients. ABC transporters are known to be involved in the efflux of antibiotics from bacterial cells (Seeger and van Veen, 2009). Since bacteria forming biofilms are encased in a matrix of extracellular polymeric substances, which render them significantly more antibiotic-resistant (Stewart and Costerton, 2001; Wu et al., 2015), it is evident that the IPF lung microbiome emphasizes key antimicrobial resistance pathways.
Beyond examining bacteria in BAL samples, a study by Kitsios et al. sought to directly analyze the microbiome of fibrotic lung tissue taken from 40 end-stage IPF patients (Kitsios et al., 2018). Contrary to previous findings, the authors found little bacterial DNA in the samples of patients with severe or acute exacerbation of IPF, therefore, comparable to controls. However, it is worth mentioning that the samples used in this study, which targeted subpleural lung tissues with significant honeycombing, originated from regions that may be unconducive to bacterial growth. Moreover, the disparity in findings may also be attributable to the end-stage sample population used in this study, compared with the early-IPF patients of most BAL studies. While the established associations between bacterial infection, progression, and severity of pulmonary fibrosis are promising, causal mechanisms have yet to be uncovered. As such, the efficacy of potential antimicrobial therapies and applications of the lung microbiome as a prognostic biomarker need to be further elucidated (Fastres et al., 2017; Ntolios et al., 2021).
4.2 Gram-positive bacteriaIn hospitalized patients with IPF, the prevalence of bacterial pneumonia, pulmonary hypertension, and lung cancer was 9.5%, 4.6%, and 3.7%, respectively (Oda et al., 2018). Among patients with bacterial pneumonia, the two most common Gram-positive pathogens were Streptococcus pneumoniae (31.6%) and methicillin-resistant Staphylococcus aureus (MRSA) (18.4%). Respiratory comorbidities, especially bacterial pneumonia and lung cancer, influence mortality in hospitalized patients with IPF (Oda et al., 2018). Notably, a recent meta-analysis revealed that bacterial streptococcal infection occurred in 99.5% of patients with IPF (Mostafaei et al., 2021). This section will discuss the relevant research regarding bacterial infection in IPF.
Streptococcus pneumoniae infection has been shown to exacerbate lung fibrosis in mice via the release of the cytotoxin pneumolysin (Knippenberg et al., 2015); notably, fibrosis progression was mitigated when mice were given a protein-based vaccine presenting a non-cytotoxic pneumolysin derivative. This suggests that pneumolysin may, in the future, be a potential target for fibrosis treatment in human patients through a protein-based pneumococcal vaccination targeting key virulence factors like pneumolysin, which could have significant preventive effects on S. pneumoniae-induced fibrosis exacerbation. However, some IPF patients undergo treatment with corticosteroids and immunosuppressive agents, which have been shown to interfere with their response to the pneumococcal vaccine (Kuronuma et al., 2018). A recent study by Bormann et al. (Bormann et al., 2021) highlighted a Cox2-dependent anti-inflammatory role of prostaglandin E2 (PGE2) in the progression of experimental pulmonary fibrosis in mice. Streptococcus pneumoniae-induced IPF progression was associated with increased expression of PGE2, and intratracheal application of PGE2 worsened fibrosis in mice with AdTGF-β1-induced lung fibrosis.
A recent study reported that Staphylococcus nepalensis is responsible for releasing a peptide, corisin, which induces apoptosis of lung epithelial cells (D’Alessandro-Gabazza et al., 2020). The study showed that mice exposed to corisin-harboring S. nepalensis experienced acute exacerbation, unlike control mice, who were either untreated or infected with corisin-free bacteria. Furthermore, human IPF patients with acute exacerbation have notably higher counts of lung corisin levels than IPF patients without exacerbation.
Further, mice infected with MRSA were found to have a more difficult time fighting off infection due to fibrosis (Warheit-Niemi et al., 2022). Specifically, the authors reported that fibrosis diminished neutrophil elastase release and oxidative radical production, inhibiting intracellular killing of MRSA by neutrophils. Not only did the fibrotic mice exhibit inhibited neutrophil activity, but lung macrophages were also shown to have a reduced capacity for phagocytosis of MRSA. Thus, the study provides evidence for impaired immune response in fibrotic lungs and proposes a mechanism for why bacterial infection in individuals with IPF increases morbidity and mortality. Overall, there has been growing evidence showing an association between Gram-positive bacteria and the development of IPF and specific mechanisms by which bacteria are responsible for exacerbating the disease and hindering the immune system’s ability to fend off infection.
4.3 Gram-negative bacteriaRespiratory infections caused by Gram-negative bacteria in IPF patients are relatively frequent during hospitalization and are reportedly effective in predicting mortality (Yamazaki et al., 2016). In 2016, a retrospective study analyzed causative pathogens in 48 IPF patients who had been hospitalized for pulmonary infections (Yamazaki et al., 2016). The study found causative pathogens in 20/48 patients, the most common of which were H. influenzae (14.5%), P. aeruginosa (4.1%), Moraxella (Branhamella) catarrhalis (4.1%), and Klebsiella pneumoniae (4.1%). Intriguingly, the causative pathogens were primarily Gram-negative bacteria, in contrast to the perspective that infection with Gram-positive bacteria causes most cases of bacterial pneumonia. Moreover, the Pneumonia Severity Index (PSI) score upon admission was significantly correlated with 30-day and hospital mortality.
Besides COPD cases in adults and more pronounced cases in infants and children with cystic fibrosis, a limited connection has been made between Bordetella pertussis and IPF, such as a 2019 case report showing that 2 patients with IPF had been diagnosed with acute exacerbation of the disease and an acute pertussis infection (Paddock et al., 2008; Bos et al., 2011; Hashemi et al., 2015; Karamooz et al., 2018; Hirai et al., 2019). Although pertussis is preventable by vaccine, neither patient had been previously vaccinated. Thus, the study suggests further investigation into pertussis as a factor in the exacerbation of IPF and the potential use of antibiotics to treat IPF patients with infection. A 2007 study investigated the role of Chlamydophila (Chlamydia) pneumoniae infection in exacerbating IPF (Tomioka et al., 2007). Twenty-seven IPF patients were tested for C. pneumoniae antibodies, IgG, and IgA, ultimately revealing that when patients presented with an acute exacerbation of IPF, C. pneumoniae was not typically present.
A recent study examined the significance of macrophage scavenger receptor 1- (MSR1)-positive cells in the progression of IPF by analyzing lung transplantation tissue samples via immunohistochemistry (Zheng et al., 2021). MSR1-positive macrophages correlated with reduced lung function and poor prognostic outcomes in IPF patients (Zheng et al., 2021). MSR1 upregulation was also significantly more common in smoking patients than in non-smoking patients but also that the expression of MSR1 was significantly elevated in IPF patients infected with K. pneumoniae, corroborating the potential role of Gram-negative bacteria in the progression of IPF. MSR1 has been suggested to play a vital role in inflammatory, innate, and adaptive immune responses, and silica-induced fibrosis (de Winther et al., 2000; Beamer and Holian, 2005).
5 Role of Haemophilus influenzae in IPFAmong the Gram-negative bacteria meriting further investigation is H. influenzae (H), as it is associated with several pulmonary afflictions. Specifically, non-typeable H. influenzae (NTHi) is a strain of Haemophilus bacteria that lacks a polysaccharide capsule, rendering it more difficult for the immune system to recognize and effectively defend against.
Several studies have outlined the role of NTHi in the development of neutrophilic asthma (Zhang et al., 2020), and others have examined the pathogenesis of NTHi infections in chronic suppurative lung diseases (Chatziparasidis et al., 2023). Moreover, extensive research has been done to explicitly investigate the role of NTHi in the pathogenesis of chronic obstructive pulmonary disease (COPD). The World Health Organization ranked COPD as the third leading cause of death in the world, with an estimated 3.23 million deaths in 2019 (Guarascio et al., 2013; World Health Organization (WHO), 2020). It is generally agreed upon that NTHi persists as one of the most common bacterial infections in adults with COPD and one of the major pathogens responsible for exacerbating the disease (Ahearn et al., 2017; Su et al., 2018). In patients with end-stage lung disease, H. influenzae was detected in the bronchi, bronchioles, damaged epithelium, and subepithelial spaces. It is thought that the near-ubiquitous presence of H. influenzae throughout the lung may function as a reservoir and facilitator of persistent infection. Intriguingly, the bacteria were observed to exist primarily extracellularly rather than in intracellular spaces (Moller et al., 1998). One study addressing H. influenzae distribution in lung tissues of patients with COPD, cystic fibrosis (CF), IPF, and other pulmonary diseases found corroborating evidence of effective and widespread invasion of pulmonary spaces. In CF, NTHi is proposed to colonize lungs early and contribute to disease pathogenesis by inciting airway epithelial inflammatory responses. It persists in part through the formation of biofilms, which one study observed in the BAL fluid of young, asymptomatic CF patients (Oda et al., 2018). NTHi may also contribute significantly to acute exacerbations in this population (Mostafaei et al., 2021). Current antibiotic treatments for CF consist primarily of P. aeruginosa-targeting tobramycin, colistin, and aztreonam (Knippenberg et al., 2015). Concerning IPF, a previously mentioned study found that Haemophilus, Streptococcus, Neisseria, and Veillonella species were associated with the disease by investigating the BAL of 65 patients; notably, they reported a 3.4-fold increase in Haemophilus in the BAL of patients with IPF in comparison to the controls (Molyneaux et al., 2014). A recurring line of reasoning has been that inflammation plays a crucial role in the pathogenesis and progression of IPF and viral and bacterial infections, which induce or worsen the condition (Bringardner et al., 2008; Homer et al., 2011). NTHi has been linked to the upregulation of pro-inflammatory pathways (Watanabe et al., 2004; Wilson et al., 2010; Yang et al., 2019).
Non-typeable Haemophilus influenza encodes several proteins that bind to plasminogen or extracellular matrix (ECM) components to damage epithelial barriers and promote persistent infection. NTHi enolase is particularly interesting, as it has been shown to bind primarily to plasminogen to manipulate plasmin’s proteolytic effects for tissue invasion (Osorio-Aguilar et al., 2021; Osorio-Aguilar et al., 2023). There is evidence of its ability to interact with the ECM components laminin, fibronectin, and collagen, and interaction with these proteins disrupts the regulation of cell-cell adhesion and migration. While the Haemophilus adhesive transporter domains (Haps) released from the proteolytic degradation of autotransporter adhesin and outer membrane lipoprotein (P4) can communicate with these elements, binding with vitronectin by P4-expressing NTHi is associated with the development of serum resistance (Fink et al., 2002; Su et al., 2016). Haemophilus surface protein E binds to both vitronectin and plasminogen to facilitate increased immune evasion (Barthel et al., 2012).
5.1 Animal studies involving NTHiSeveral studies have revealed the importance of cytokines in NTHi. For example, interleukin-17A, a pro-inflammatory cytokine, has been demonstrated to play a role in lung fibrosis in the context of bleomycin-induced fibrosis in mice (Wilson et al., 2010). In addition, murine model of bleomycin-induced lung fibrosis has found that dysregulated lung microbiota can promote the production of interleukin-17B (IL-17B), driving disease progression (Yang et al., 2019). Other studies utilizing similar models to simulate bacterium-induced acute exacerbation of IPF by administering bleomycin to mice and then infecting them with a strain of NTHi (Chen et al., 2022) have revealed that NTHi infection can cause acute exacerbation of IPF and that the IL-17 gene is key for the progression of IPF acute exacerbation and could serve as a novel therapeutic target for treating the disease.
The decline in pulmonary function and development of fibrotic lung tissue is heavily associated with localized lung tissue inflammation (Bringardner et al., 2008; Homer et al., 2011). Several other pro-inflammatory cytokines, such as interleukin-8, interleukin-1 beta, and chemokine (C-X-C motif) ligand 1, are known to drive localized inflammation (Harada et al., 1994; Dinarello, 2011; Donahoe et al., 2015; Sawant et al., 2015). NF-kB is a transcription factor that has multiple impacts on target cells, such as regulating inflammation, inducing apoptosis, and regulating cell growth (Liu et al., 2017). NTHi infection has been shown to upregulate NF-kB activation with tumor necrosis factor, a pro-inflammatory cytokine (Watanabe et al., 2004). NF-kB is documented to have pro-inflammatory effects on fibroblasts, cells responsible for secreting collagen and extracellular matrix proteins, through regulation of gene transcription (Htwe et al., 2015). Fibroblast stimulation to secrete collagen and extracellular matrix proteins is one of the primary mechanisms that lead to the development of pulmonary fibrosis (Hanmandlu et al., 2022). As fibroblasts secrete increased amounts of collagen and extracellular matrix proteins, lung tissue hardens and interferes with proper gas exchange. NF-kB has also been shown to stimulate fibroblasts to secrete pro-inflammatory cytokines such as interleukin-8, macrophage inflammatory protein-1-alpha, and transforming growth factor-beta (Htwe et al., 2015). Moreover, a 2017 study found that lung fibroblasts could internalize live NTHi, and that fibroblasts were responsible for activating IFN-γ and IL-17A cytokine production via autologous NTHi-specific lung CD4+ T cells. This suggests that human lung fibroblasts play a crucial role in mediating T helper (Th) cell responses to bacterial infection, and specifically in conjunction with NTHi (Hutton et al., 2017).
Macrophages have also been shown to respond to NF-kB to secrete pro-inflammatory cytokines (Liu et al., 2017). Given that macrophages are present within alveoli, this could be another driving mechanism for localized inflammation. NF-kB is also documented to stimulate fibroblast transformation into myofibroblasts, a fibroblast state that secretes increased amounts of extracellular matrix proteins (Liu et al., 2017). Mice treated with NF-kB inhibitors were significantly protected from lung fibrosis development compared to control mice (Thakur et al., 2022). Given the wide range of effects that NF-kB has been shown to induce in various cell types, future research regarding NTHi and NF-kB upregulation could provide valuable information for pulmonary fibrosis pathogenesis.
Numerous mechanisms that enable NTHi to not only establish itself in patients with IPF, but also to maintain a persistent infection for prolonged periods of time are discussed (Murphy et al., 2004). Future studies will further reveal the specific mechanisms of H. influenzae in driving pathogenesis in individuals with IPF, using previously established research on related illnesses such as COPD.
5.2 Mechanisms involved in NTHi pathogenesisNTHi has adapted several strategies to avoid the host immune response and maintain a persistent infection. For instance, NTHi uses a variety of adhesins, which allow the bacteria to attach itself to and invade host cells (Duell et al., 2016). In essence, these adhesins are essential in permitting NTHi to colonize primary sites, allowing further infection in secondary sites and eventually leading to the formation of biofilms and mediation of key virulence mechanisms (Duell et al., 2016). Biofilms comprise an extracellular polymeric substance inhabited by a community of bacteria. NTHi produces these protective biofilms to maintain a reserve of bacteria which can go on to cause subsequent infections (Gunn et al., 2016). Thus, the formation of biofilms is a key mechanism by which NTHi and other pathogens can sustain persistent infections in the host.
Studies have revealed that NTHi does not actually bind to the cell surface directly, but rather adheres to host vitronectin by means of an outer membrane protein, protein-E. The protein-E-vitronectin interaction plays a role in the adherence and invasion of NTHi in bronchial epithelial cells. It could serve as a potential target for novel treatments and vaccines (Ikeda et al., 2015). Lysostaphin-like metalloproteases (LytM proteins), known to facilitate cell division by affecting cleavage and membrane composition, contributed to the pathogenesis and physiology of NTHi. Data suggests explicitly that the component of outer membrane and cell wall physiology, the murein hydrolase activator, EnvC protein, possessing a LytM domain, might impact bacterial surface protein composition via a mechanism in which EnvC facilitates the transport of periplasmic chaperones to the outer membrane. This is further supported by studies showing that an NTHi envC-defective strain has diminished capacity to adhere to epithelial cells and form biofilm, while also displaying a reduced resistance to the immune system (Ercoli et al., 2015).
Although NTHi is widely considered an extracellular pathogen, NTHi has also been spotted residing intracellularly (St Geme and Falkow, 1990). For example, NTHi lipooligosaccharide interacts with the platelet-activating factor receptor, enabling NTHi to adhere to and infiltrate human bronchial epithelial cells (Swords et al., 2000). Morey et al. proposed a unique mechanism by which NTHi can invade the airway epithelium and reside intracellularly, involving the assembly of microtubules, integrity of lipid rafts, and activation of phosphatidylinositol 3-kinase (PI3K) signaling (Morey et al., 2011). NTHi were found to primarily reside in acidic sub-cellular vacuoles with late endosome features, existing in a metabolically active yet non-replicative state. This NTHi-containing vacuole co-localizes with LysoTracker, lysosome-associated membrane protein 1 (LAMP1), LAMP2, CD63, and Rab7 and does not possess Golgi- or autophagy-related markers. It may modulate epigenetic changes like many known bacterial nucleomodulins or by altering host endosomal proteins mechanisms to evade host immune response and gain intracellular persistence (Morey et al., 2011; Cohen et al., 2013; Denzer et al., 2020; Fol et al., 2020; Wrede et al., 2023).
Further investigation has suggested that NTHi strategically positions itself intracellularly to avoid immune responses and maintain a persistent infection, and IgA proteases are essential to NTHi’s ability to do this. Epithelium and airway mucous membranes contain IgA as a part of the innate immune system to defend against pathogens; in the upper respiratory tract, IgA1 is the dominant form (Salvi and Holgate, 1999). H. influenzae is documented to produce three types of IgA1 proteases that can cleave the heavy chain of IgA1 antibodies and render the Fc portion dysfunctional. While most capsulated strains of H. influenzae produce only one of the three IgA1 proteases, NTHi strains produce any of the three types (Foxwell et al., 1998). Each of the three proteases play distinct roles in the pathogenesis of NTHi. While IgA protease is necessary for NTHi invasion, IgA proteases B1 and B2 are essential to the intracellular persistence of NTHi (Murphy et al., 2017). Not only do IgA proteases serve to protect NTHi from mucosal immunity by cleaving human IgA1, but they have also been found to facilitate intracellular survival by cleavage of LAMP1 at each stage of the endolysosomal pathway, including the plasma membrane, early and late endosome, and lysosome stages. In doing so, IgA proteases B1 and B2 facilitate NTHi’s ability to evade the endo-lysosomal pathway, though NTHi is ultimately destroyed in lysosomes after variable durations of intracellular survival (Clementi et al., 2014). NTHi has also been documented invading epithelial cells and residing in membrane-bound vacuoles inside epithelial cells (Foxwell et al., 1998). The mechanisms of NTHi pathogenesis are depicted in Figure 2.
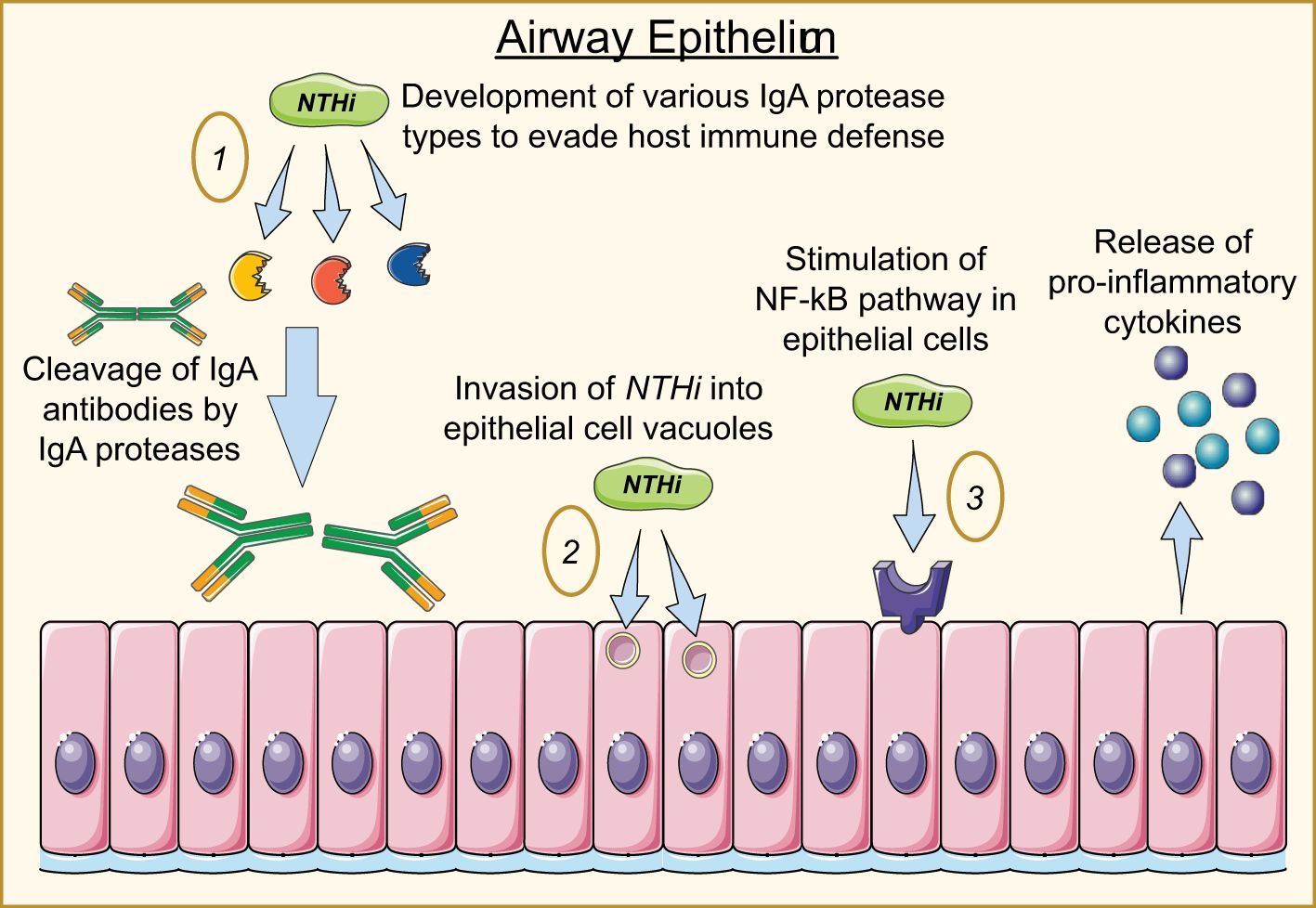
Figure 2. Mechanisms of NTHi invasion into airway epithelial cells. 1) NTHi produces three IgA protease types responsible for destroying IgA antibodies and enabling the bacterium to invade and persist within the host cell. 2) NTHi enters the cell through endocytic mechanisms, hiding in vacuoles to avoid degradation. 3) NTHi stimulates the NF-kB pathway, producing cytokine secretion while promoting a series of pro-inflammatory responses from fibroblasts and macrophages.
To ensure that NTHi persists in various hostile COPD environments, the bacteria use several mechanisms for genetic variation, including antigenic variation (Sethi et al., 2004), phase variation (Murphy et al., 2004), and epigenetic variation (Atack et al., 2015). Additionally, NTHi must adapt specific metabolic pathways to take in necessary nutrients. For instance, ferric uptake regulator (Fur)-regulated genes in NTHi modulate iron usage within the cell, ultimately facilitating more severe and persistent infections (Duell et al., 2016).
Because iron is in limited supply within the host, NTHi sequesters iron to ensure its survival. Recent dual RNA-Seq studies have further elucidated these metabolic adaptations, revealing that NTHi upregulates iron acquisition pathways and modifies metabolic processes to survive under intracellular conditions (Baddal et al., 2015). These studies also show that NTHi dynamically adjusts its transcriptional responses in real time, responding to the intracellular environment to maximize survival. Concurrently, NTHi modulates host immune responses, suppressing inflammatory signaling pathways to evade detection and maintain a permissive intracellular environment (Ackland et al., 2021). This dual role of metabolic adaptation and immune modulation enables NTHi to persist intracellularly, protecting itself from immune clearance and ensuring long-term colonization of the host.
Using the mechanisms outlined above, NTHi can establish a persistent infection in the lower airways, lasting months to years. Moreover, this wide range of unique virulence mechanisms allows NTHi to persist in its host and exacerbate pulmonary diseases, including but not limited to IPF. Therefore, NTHi is particularly interesting when evaluating possible concurrence and associations with IPF. Mechanisms discussed above could play a role in the progression of IPF in patients infected with H. influenzae.
5.3 Limitations of bleomycin model of pulmonary fibrosis in preclinical studiesMost preclinical lung fibrosis studies widely use the chemotherapy drug bleomycin sulfate, which is intratracheally administered in rats and mice to induce an inflammatory response progressing into lung fibrosis that mimics the pathology of IPF and airway remodeling (Polosukhin et al., 2012; Tanjore et al., 2013). Bleomycin-induced pulmonary fibrosis is highly reproducible, with key molecular pathways triggered by reactive oxygen species and inflammation that leads to excessive collagen deposition and fibrosis (Degryse et al., 2011; Polosukhin et al., 2012; Tanjore et al., 2013; Moss et al., 2022). The rodent models are also employed to examine the in vivo efficacy of drugs for preventive and therapeutic potentials for IPF treatment (Kolb et al., 2020). While these models capture many qualitative histological aspects of IPF, they may not completely replicate all the complexities of the human disease, given the chronic nature of the disease in IPF patients. On a histologic level, bleomycin-induced fibrosis lacks the basal and subpleural predominance characteristic of IPF and involves more limited alveolar epithelial cell remodeling (Moss et al., 2022). However, repetitive bleomycin dosing and dosing of aged mice show promising results that more closely approximate non-reversible fibrotic phenotypes and classic histologic presentations of IPF (Degryse et al., 2010; Weckerle et al., 2023). Interestingly, positron-emission tomography-computed tomographic scanning was employed in bleomycin-induced mice to closely mimic the diagnostic methods of IPF and evaluate pathologic changes with different modes of bleomycin administration. It was shown that repeated intravenous bleomycin delivery for seven consecutive days matched closer to the early pathologic features of idiopathic pulmonary interstitial fibrosis (Sgalla et al., 2016; Comes et al., 2020; Gul et al., 2023).
6 Antibiotics and other therapeutics in the treatment of IPF6.1 Completed clinical trials6.1.1 AntiviralsNumerous studies have evaluated the use of antibiotics to help treat IPF. Despite the prevalence of IPF worldwide, our understanding of how persistent infection plays into the pathogenesis and advancement of the illness is still incomplete. Much of the research on viral infections has centered on the herpesviruses, particularly EBV. Although the presence of EBV in IPF patients is valid, little research has been done to investigate the potential role of other viruses, such as human metapneumovirus, influenza virus, and coronaviruses. Exploring the comorbidity of other viruses with IPF is beneficial, especially considering that evidence points to antiviral regimens as a valid approach to treating the disease. Ganciclovir, an antiviral medication, successfully mitigates the progression of IPF (Folcik et al., 2014), and concurrent treatment with pirfenidone, the anti-fibrotic drug, and valganciclovir, the prodrug of ganciclovir, was well tolerated in patients with IPF and could serve as a viable treatment (Study# 1, Table 1) (Blackwell et al., 2021). Azithromycin, in addition to its antimicrobial properties, has been found to possess anti-inflammatory effects, modulating the immune response by reducing the production of inflammatory cytokines and chemokines (Gao et al., 2010). Like other macrolide antibiotics, azithromycin can also penetrate cells and accumulate intracellularly, which is particularly relevant in treating intracellular bacterial infections such as those discussed for NTHi. In fact, Azithromycin’s capacity for intracellular activity may contribute to its anti-inflammatory properties, as it can modulate immune responses within cells (Munic et al., 2011). A retrospective study assessing the effects of azithromycin found that the mortality rate in IPF patients treated with azithromycin (26%) was significantly lower than that of those treated with fluoroquinolones (70%) (Kawamura et al., 2017). A more recent retrospective study investigated how IPF patients responded to prophylactic azithromycin. This study found that hospital admissions and antibiotic courses were significantly lower in the 12 months following the use of prophylactic azithromycin compared to the no-prophylactic treatment group (Macaluso et al., 2019). However, a 2021 randomized controlled trial showed no substantial benefit from treatment with low-dose azithromycin for chronic cough in patients with IPF (Study #3, Table 1) (Guler et al., 2021). The most prominent and recent SARS-COV2-associated lung disease, COVID-19, shares common risk factors with IPF. These risk factors include age-associated inflammation and metabolic syndromes, particularly in male patients.
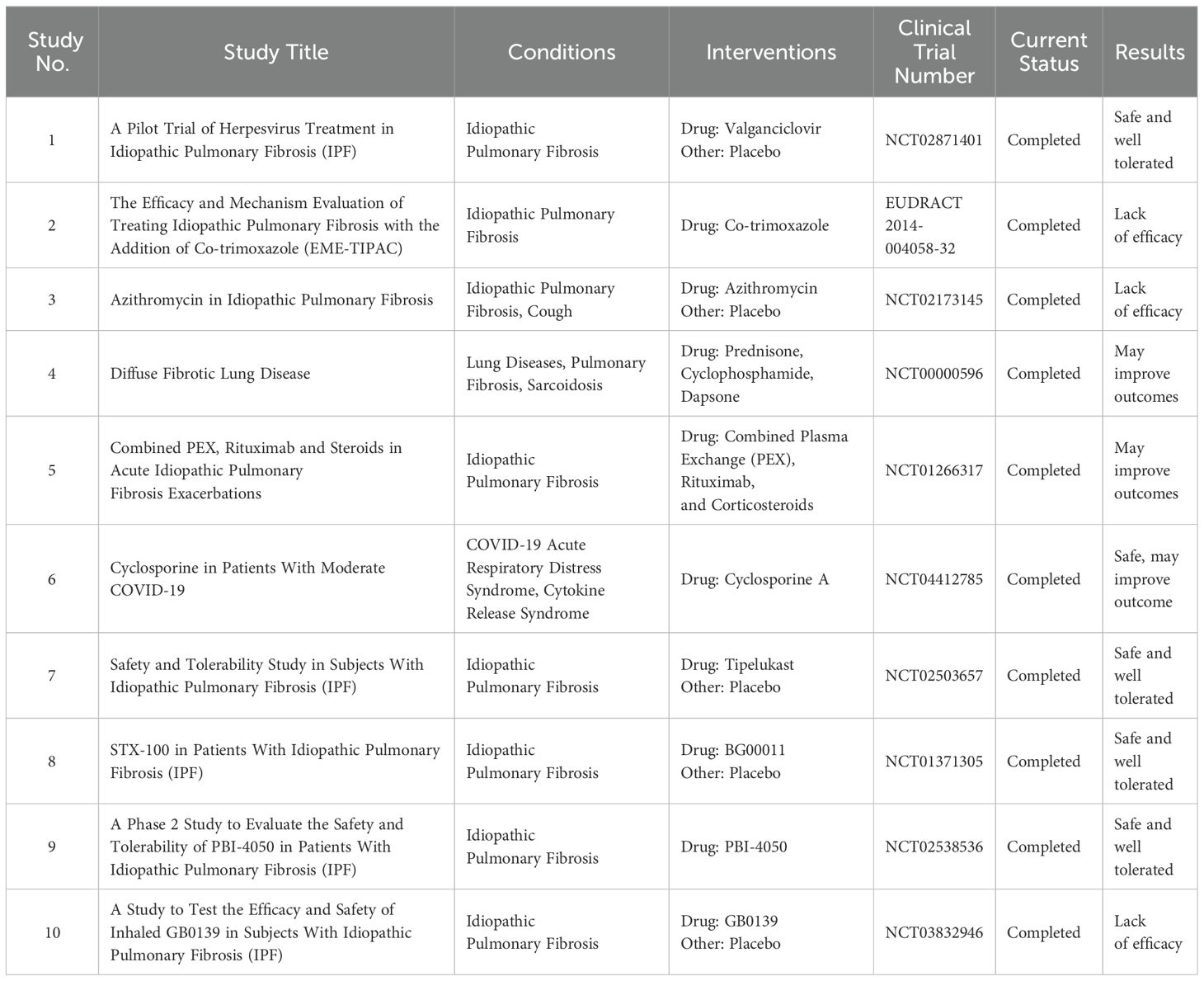
Table 1. List of completed clinical trials conducted in IPF patients.
Thus, available antifibrotic therapies could help prevent severe COVID-19 in patients with IPF or treat severe COVID-19 in patients without IPF (George et al., 2020). The safety and effectiveness of cyclo
留言 (0)