
記住我


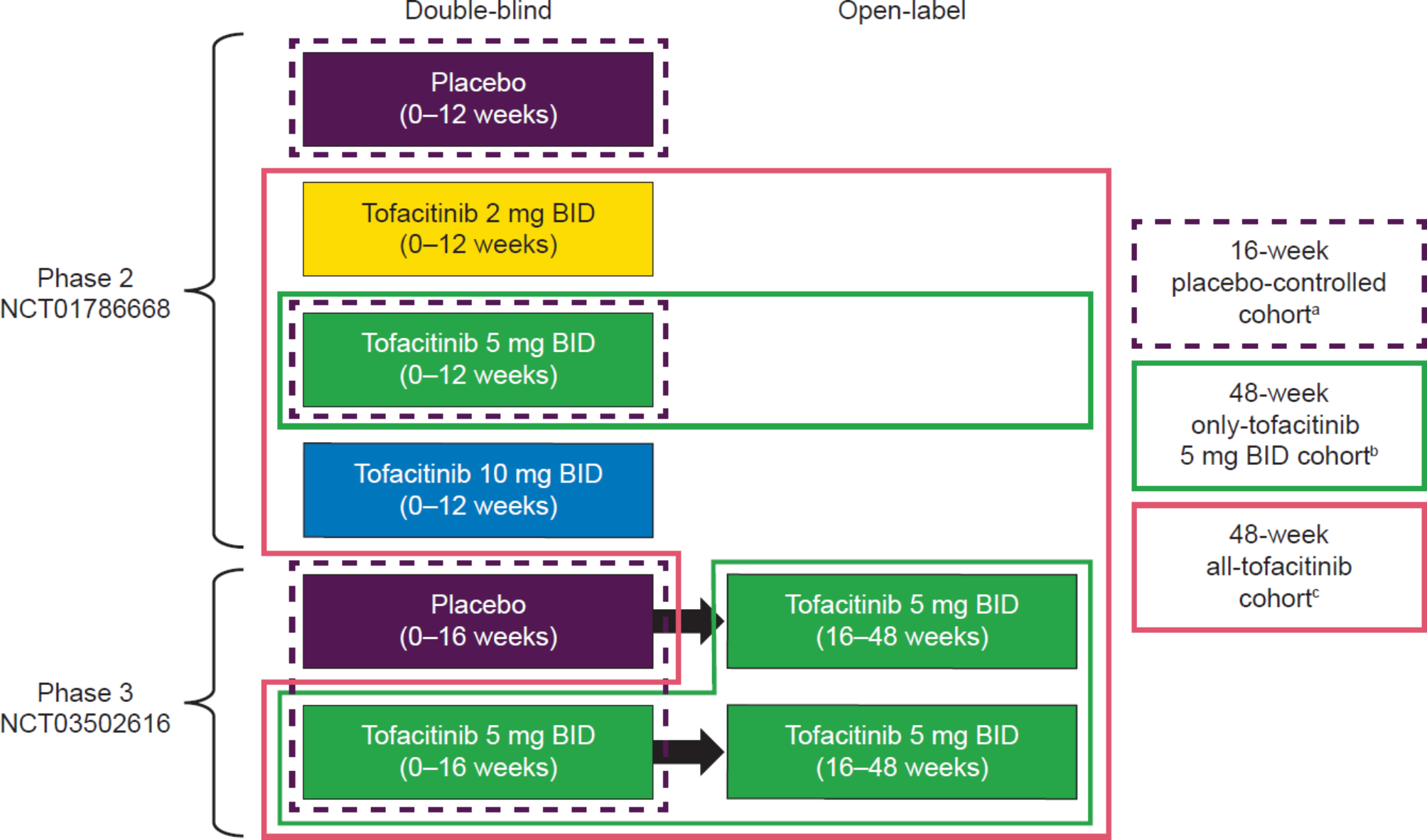





Overall, 372, 316, and 420 patients with AS were in the 16-week placebo-controlled cohort, and 48-week only-tofacitinib 5 mg BID and all-tofacitinib cohorts, respectively. Baseline characteristics were generally similar between tofacitinib 5 mg BID and placebo (16-week placebo-controlled cohort), and consistent with the 48-week all-tofacitinib cohort (Table 1).
Table 1 Baseline demographics and disease characteristicsa for patients in tofacitinib AS, RA, and PsA safety cohortsAt baseline, 3.5% and 3.6% of patients in the 48-week only-tofacitinib 5 mg BID and all-tofacitinib cohorts, respectively, had HxASCVD; in patients without HxASCVD, > 75% had low (< 5%) baseline 10-year ASCVD risk (Fig. 2; Supplementary Table 2 in Additional File 2).
Fig. 2
Baseline CV risk profile of patients in the tofacitinib AS Phase 2/3 safety cohorts. HxASCVD was defined as ≥ 1 occurrence of CAD, CeVD, or PAD. History of CAD, CeVD, or PAD was identified in patients’ general medical history through MedDRA Preferred Terms consistent with one of these conditions and reflecting prior/ongoing events, procedures, or diagnoses. Percentages were based on all patients as the denominator (n = 316 for 48-week only-tofacitinib 5 mg BID cohort; n = 420 for 48-week all-tofacitinib cohort). Baseline 10-year ASCVD risk was determined using the ASCVD-PCE risk calculator, developed by the American College of Cardiology/American Heart Association [19]. Scores were calculated based on patients’ age, sex, race (White, Black, other), smoking status (yes/no), systolic blood pressure, antihypertensive treatment (yes/no), total cholesterol, high-density lipoprotein cholesterol, and diabetes (yes/no). Percentages based on all patients with nonmissing values. Patients without HxASCVD but with missing ASCVD-PCE risk data: 48-week only-tofacitinib 5 mg BID cohort, n = 1; 48-week all-tofacitinib cohort, n = 2. AS ankylosing spondylitis, ASCVD atherosclerotic cardiovascular disease, BID twice daily, CAD coronary artery disease, CeVD cerebrovascular disease, Hx history (of), MedDRA Medical Dictionary for Regulatory Activities, N total number of patients included in analysis, n number of patients with characteristic, PAD peripheral artery disease, PCE pooled cohort equations, RCT randomized controlled trial
RA Phase 2/3 and PsA Phase 3 safety cohortsThe RA Phase 2/3 and PsA Phase 3 safety cohorts included 2664 and 347 patients, respectively; at baseline, patients were older vs. the AS Phase 2/3 safety cohorts, and included greater proportions of concomitant csDMARD and oral corticosteroid users, and fewer males and current smokers (Table 1).
AS real-world cohortThe AS real-world cohort comprised 2253 patients; 39.5% had received prior bDMARDs (Supplementary Table 3 in Additional File 2). At baseline, vs. the AS Phase 2/3 safety cohorts, there was a greater proportion of females and patients with prior oral corticosteroid use.
Overview of AEs in the AS Phase 2/3 safety cohortsIn the 16-week placebo-controlled cohort, proportions of patients with TEAEs, serious AEs, severe AEs, and discontinuations due to AEs were similar between tofacitinib 5 mg BID and placebo (Table 2). Uveitis was reported in one (0.5%) and three (1.6%) patients with history of uveitis receiving tofacitinib 5 mg BID and placebo, respectively. Psoriasis occurred in one (0.5%) patient (placebo) with history of psoriasis.
Table 2 Summary of AEs in the tofacitinib AS phase 2/3 safety cohortsaIn the 48-week cohorts, < 4% of patients reportedserious AEs, severe AEs, and discontinuations due to AEs (Table 2). Four (1.3%) and six (1.4%) patients in the 48-week only-tofacitinib 5 mg BID and all-tofacitinib cohorts, respectively, experienced uveitis; all but one patient (tofacitinib 2 mg BID) had history of uveitis.
No TEAEs of inflammatory bowel disease occurred; most TEAEs were mild/moderate. No deaths were reported.
Safety event IRs by age, sex, race, geographic region, prior bDMARD history, and Day 1 concomitant csDMARD use, in the 48-week cohorts are shown in Supplementary Tables 4–9 in Additional File 3 (noting the small numbers of patients in some subgroups and overlapping CIs for some comparisons).
AEs of special interest in the AS Phase 2/3 safety cohorts, RA Phase 2/3 and PsA Phase 3 safety cohorts, and AS real-world cohortSerious infectionsOne patient receiving tofacitinib 5 mg BID (included in all AS Phase 2/3 safety cohorts) had a serious infection (aseptic meningitis [Table 3]). No serious infections were reported with placebo.
Table 3 General AEs and AEs of special interest in tofacitinib AS, RA, and PsA safety cohortsSerious infection IRs (95% CIs) were 1.77 (0–5.89) vs. 0 (0–3.31) per 100 PY for the tofacitinib 5 mg BID vs. placebo arms (16-week placebo-controlled cohort). Serious infection IRs (95% CIs) were 0.43 (0.01–2.41) and 0.38 (0.01–2.12) per 100 PY for the 48-week only-tofacitinib 5 mg BID and all-tofacitinib cohorts, respectively; these were numerically lower vs. the RA Phase 2/3 and PsA Phase 3 safety cohorts (Table 3), and the AS real-world cohort (Supplementary Table 10 in Additional File 4), with 95% CIs mostly overlapping.
Opportunistic infectionsNo opportunistic infections were reported in the AS Phase 2/3 safety cohorts (Table 3); three events were assessed by an independent adjudication committee (aseptic meningitis and ophthalmic herpes simplex [tofacitinib 5 mg BID]; herpes zoster [tofacitinib 10 mg BID]), but did not meet opportunistic infection criteria. Across other cohorts, opportunistic infections IRs were ≤ 0.5 per 100 PY, with overlapping 95% CIs (Table 3; Supplementary Table 10 in Additional File 4).
TuberculosisNo tuberculosis cases were reported in the AS Phase 2/3 safety cohorts, PsA Phase 3 safety cohort (Table 3), or AS real-world cohort (Supplementary Table 10 in Additional File 4); tuberculosis IR was < 0.1 per 100 PY for the RA Phase 2/3 safety cohort (Table 3).
Herpes zosterHerpes zoster (HZ) occurred in five (1.6%) and seven (1.7%) patients in the 48-week only-tofacitinib 5 mg BID and all-tofacitinib cohorts, respectively (Table 3); all cases involved a single dermatome, except for one patient (tofacitinib 10 mg BID) with HZ involving two adjacent dermatomes (this did not meet opportunistic infection criteria on adjudication). All cases of HZ were non-serious.
HZ IRs (95% CIs) were 2.18 (0.71–5.08) and 2.68 (1.08–5.53) per 100 PY for the 48-week only-tofacitinib 5 mg BID and all-tofacitinib cohorts, respectively (Table 3). These were similar vs. the RA Phase 2/3 and PsA Phase 3 safety cohorts (Table 3), and numerically higher vs. the AS real-world cohort (overlapping 95% CIs [Supplementary Table 10 in Additional File 4]).
MalignanciesNo malignancies (excluding nonmelanoma skin cancer [NMSC]) or NMSCs were reported in the AS Phase 2/3 safety cohorts (Table 3). Across the RA Phase 2/3 and PsA Phase 3 safety cohorts, and AS real-world cohort, malignancies (excluding NMSC) and NMSCs occurred in 0–0.9% and 0–0.4% of patients, respectively; and IRs of malignancies (excluding NMSC) and NMSCs were all < 1.5 and < 0.5 per 100 PY, respectively, with 95% CIs overlapping (Table 3; Supplementary Table 10 in Additional File 4).
Cardiovascular eventsNo MACE, deep vein thrombosis (DVT), pulmonary embolism (PE), VTE, or arterial thromboembolism events (ATE) were reported in the AS Phase 2/3 safety cohorts (Table 3). Among the RA Phase 2/3 and PsA Phase 3 safety cohorts, and AS real-world cohort, MACE, VTE, DVT, PE, and ATE occurred in 0.2–0.4%, 0–0.3%, 0–0.2%, 0–0.1%, and 0–0.3% of patients, respectively, and IRs were all generally < 1 per 100 PY, with 95% CIs overlapping (Table 3; Supplementary Table 10 in Additional File 4).
Gastrointestinal perforationsNo gastrointestinal perforations were observed in the AS Phase 2/3 safety cohorts, RA Phase 2/3 safety cohort, or AS real-world safety cohort (Table 3; Supplementary Table 10 in Additional File 4); gastrointestinal perforation IR (95% CI) was 0.50 (0.01–2.77) per 100 PY for the PsA Phase 2/3 safety cohort (Table 3).
Liver parameters, hepatic-related AEs, and drug-induced liver injury (AS Phase 2/3 safety cohorts only)In the tofacitinib 5 mg BID arm of the 16-week placebo-controlled cohort, aspartate aminotransferase and alanine aminotransferase levels increased from baseline to week 12, then decreased to week 16, while total bilirubin levels generally increased from baseline to week 16; overall, similar trends to week 16 were seen in the 48-week cohorts, with levels stabilizing from weeks 16 to 40, then increasing to week 48 (Fig. 3). Absolute values for liver parameters over time are shown in Supplementary Fig. 1 in Additional File 5 and patients with laboratory values meeting monitoring/discontinuation criteria are summarized in Supplementary Table 11 in Additional File 5. There were no events of liver enzyme elevations that were adjudicated as probable drug-induced liver injuries.
Fig. 3
Change from baseline in AST, ALT, and total bilirubin with tofacitinib in AS Phase 2/3 safety cohorts. Baseline was defined as the last non-missing assessment prior to the first dose of investigational product (including placebo) in the 16-week placebo-controlled cohort and prior to the first dose of tofacitinib in the 48-week cohorts. Within the 16-week placebo-controlled and 48-week cohorts, patients in the Phase 2 RCT received tofacitinib to week 12. ALT alanine aminotransferase, AS ankylosing spondylitis, AST aspartate aminotransferase, BID twice daily, n total number of patients, SE standard error
Supplementary Table 12 in Additional File 5 summarizes patients with confirmed transaminase and bilirubin elevations. There were no Hy’s Law cases.
Change from baseline in laboratory values and vital signs in the AS Phase 2/3 safety cohortsHematologic parametersAbsolute values and changes from baseline for hematology parameters over time are shown in Supplementary Figs. 2 and 3 (in Additional File 6), respectively. For all AS Phase 2/3 safety cohorts, there were no study drug discontinuations due to hemoglobin reductions, neutropenia, lymphopenia, or thrombocytopenia (Supplementary Table 11 in Additional File 5).
Lipid parametersAbsolute values for total cholesterol, high-density and low-density lipoprotein cholesterol, and triglycerides over time are shown in Supplementary Fig. 4 in Additional File 6. In the tofacitinib 5 mg BID arm of the 16-week placebo-controlled cohort, levels increased to week 4, then were generally sustained to week 16; overall, similar trends were observed for the first 16 weeks of the 48-week tofacitinib cohorts, with levels sustained to week 48.
Creatine kinase and serum creatinineAbsolute values and changes from baseline for creatine kinase and serum creatinine over time are shown in Supplementary Figs. 5 and 6 (in Additional File 6), respectively. No patients met drug discontinuation criteria for creatine kinase or serum creatinine (Supplementary Table 11 in Additional File 5).
Vital signsThere were no clinically meaningful changes over time in heart rate (data not shown), electrocardiogram parameters (data not shown), or blood pressure (Supplementary Fig. 7 in Additional File 6).
Hypertension was reported by four (2.2%) and two (1.1%) patients receiving tofacitinib 5 mg BID and placebo, respectively, in the 16-week placebo-controlled cohort, and by nine (2.9%) and 11 (2.6%) patients in the 48-week only-tofacitinib 5 mg BID and all-tofacitinib cohorts, respectively (all hypertension AEs were mild/moderate).
Changes from baseline in body weight over time are shown in Supplementary Fig. 8 in Additional File 6; in the 16-week placebo-controlled cohort, greater increases in body weight were observed for tofacitinib 5 mg BID vs. placebo; generally, in the 48-week cohorts, body weight increased to week 16, then stabilized thereafter to week 48.
留言 (0)