
記住我




To investigate the expression of TRGs in HCC, we downloaded the expression data of 19,938 genes from tumors and paracancerous tissues from the TCGA database and obtained 2,093 TRGs from TelNet (Supplementary Table 1). The total number of genes differently expressed in cancer and paracancer tissues was 16902, with 2093 TRGs remaining after intersections with TRGs (Fig. 1A). The principal component analysis revealed that malignant and paracancerous tissues may be distinguished based on TRGs expression (Fig. 1B). To further analyze, 949 differential genes with significant differences in tumor and paracancerous tissues were identified based on the significant differential expression condition |logFC|> 1, fdr < 0.05 (Supplementary Table 2). The distribution of the top 100 TRGs with significant differences was presented as a heat map (Fig. 1C).
Fig. 1
Analysis of differential expression of TRGs in HCC patients. A Venn diagram showing the number of differentially expressed TRGs in cancer and paracancer tissues; B principal component analysis of HCC vs normal controls; C heatmap showing the top 100 TRGs significantly differentially expressed in HCC and normal tissues
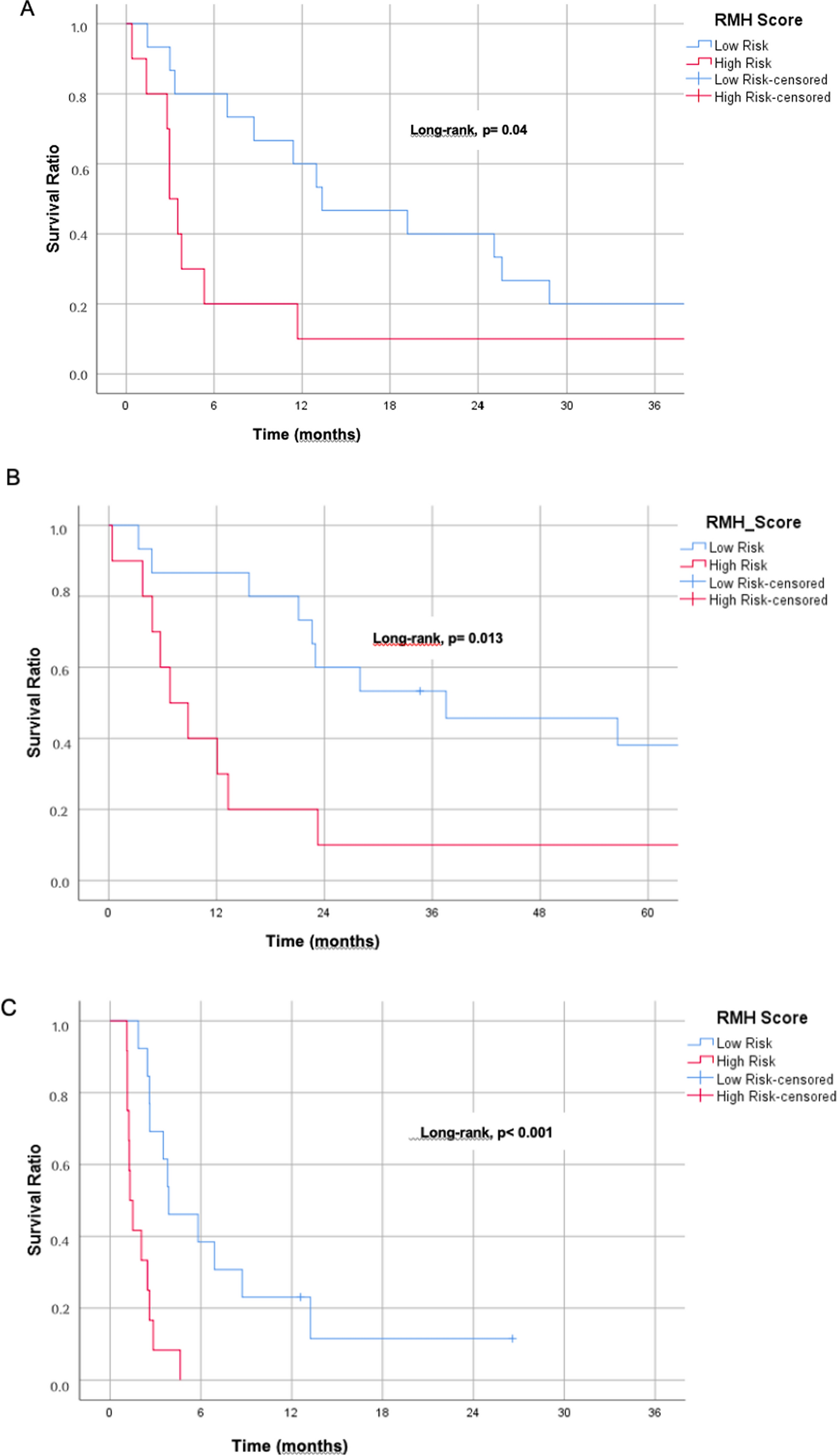
3.2 Construction and validation of a risk scoring model for TRGs based on the TCGA-HCC cohortAmong the 949 TRGs screened above that were significantly differentially expressed in HCC and paracancerous tissues, we identified 657 genes (Supplementary Table 3) that were strongly associated with prognosis using univariate COX regression analysis in conjunction with patients' clinical information (Fig. 2A). After LASSO regression analysis, we finally selected seven core differential genes to construct the risk score model: CDCA8, DTYMK, KPNA2, MRTO4, RAD54B, RRAGC and RTN3, and the risk score calculation equation was:CDCA8 × 0.02 + DTYMK × 0.004 + KPNA2 × 0.152 + MRTO4 × 0.013 + RAD54B × 0.095 + RRAGC × 0.049 + RTN3 × 0.03 (Fig. 2B). Compared to paracancerous tissues, these seven hub gene expressions were significantly elevated in tumor tissues (Fig. 2C). Patients were divided into low-risk and high-risk groups based on their median risk score. Survival analysis showed that the high-risk group had significantly worse overall survival (OS) compared to the low-risk group (P < 0.001, Fig. 2D).
Fig. 2
Construction and validation of a risk score model for HCC patients based on TRGs. A Volcano plot showing TRGs and their risk ratios significantly associated with prognosis after univariate COX regression analysis; B the optimal model was obtained by the tenfold cross-validation method of Lasso regression analysis; C box plots of 7 risk genes differentially expressed in HCC and normal tissues; D the results of the survival analysis showed differences in survival between the different risk subgroups. (*p < 0.05, **p < 0.01, ***p < 0.001,****p < 0.0001)
3.3 Construction and validation of nomograms based on risk scores and clinicopathologic indicatorsThe results of univariate and multivariate COX regression showed that the risk score model could be done as an independent predictor of prognosis in patients with HCC (Fig. 3A and B). The results of the ROC curve showed a better predictive performance of the risk score (AUC = 0.811), which was superior to the indicators of age (AUC = 0.0.492), gender (AUC = 0.508), grade (0.489) and stage (0.712) (Fig. 3C). Furthermore, the survival prediction AUC values of this prediction model for 1, 3, and 5 years were 0.811, 0.697, and 0.643, indicating that this risk scoring model performed well (Fig. 3D). Nomograms are appropriate for joint diagnosis or prediction of disease onset and progression using numerous indicators, and they can be used to assess the effectiveness of prediction models. To further evaluate the model's validity and accuracy, we created nomograms, and the results revealed that the model could accurately predict the 1-, 3-, and 5-year prognosis probability of HCC patients (Fig. 3E). The calibration curves show good predictive performance of the model for 1, 3 and 5 years (Fig. 3F).
Fig. 3
Construction of nomograms based on the TCGA-HCC cohort and validation of their clinical efficacy. A and B Univariate COX regression analysis and multivariate COX regression analysis were used to screen for independent influences associated with prognosis; C ROC curves for prognostic predictive efficacy of clinicopathologic features and risk score models; D ROC curves for 1-, 3- and 5-year predictive efficacy of risk scoring models; E nomograms constructed based on clinicopathologic features and risk scoring models; F calibration curves for 1-, 3-, and 5-year predicted efficacy of nomograms
3.4 Correlation between the risk score model constructed on the basis of 7 TRGs and immune infiltrationTumor immune microenvironment is closely related to the therapeutic efficacy and prognosis of HCC patients, so we next evaluated the infiltration of 22 immune cells in the high- and low-risk groups, in which five types of immune cell infiltration were significantly different, including T cells CD4 memory resting (P < 0.01), T cells CD4 memory activated (P < 0.05), NK cells activated (P < 0.05), Monocytes (P < 0.001) and Macrophages M0 (P < 0.01) five immune cell infiltrations were significantly different (Fig. 4A). The results of the correlation of hub genes and risk scores with immune cells were also presented as a heat map (Fig. 4B). Further analysis of the differences between immunity-rich subtypes in patients with different risk groups showed that the proportion of high-risk patients with C1(10%), C2 (18%), and C4 (46%) was higher compared to those with low risk groups, and that the ratio of C3 (26%) was lower compared with those with lower risk (51%). The difference in immune subtype enrichment was significant between the two subgroups (P < 0.001) (Fig. 4C). In the present study, we found that patients in the high-risk group had higher TIDE scores and that tumors may achieve immune escape by suppressing the activity of cytotoxic T lymphocytes (Fig. 4D).
Fig. 4
Differences in the tumor immune microenvironment between risk subgroups. A Differences in the abundance of 22 immune cell infiltrations in the tumor immune microenvironment of HCC patients in the high- and low-risk groups; B heatmap of the correlation of risk scores and the seven risk genes they contain with immune cells; C differences in immune subtype enrichment between risk groups; D distribution of TIDE scores. (*p < 0.05, **p < 0.01, ***p < 0.001,****p < 0.0001)
3.5 Risk score could predicts the treatment strategyExploring the correlation between risk scores and drug therapy showed that risk scores were significantly correlated with sensitivity to some drugs. Sorafenib (r = 0.31, P = 5 × 10–9), mitoxantrone (r = 0.28, P = 1.2 × 10–7), oxaliplatin (r = 0.29, P = 7.7 × 10–8), gemcitabine (r = 0.33, P = 3.9 × 10–10) and entinostat (r = 0.28, P = 2.1 × 10–7) were more sensitive when the risk score was higher, while vincristine (r = −0.3, P = 2.3 × 10–8), cyclophosphamide (r = −0.22, P = 4.5 × 10–5), paclitaxel (r = −0.45, P = 2.2 × 10–16) and 5-Fluorouracil (r = −0.38, P = 3.5 × 10–13) showed a gradual decrease in sensitivity as the risk score increased (Fig. 5A-I), and the results of the above study may provide guidance for clinical drug therapy in HCC.
Fig. 5
Therapeutic sensitivity of targeted drugs. A–I Correlation between IC50 values and TRG risk scores for the top 9 targeted therapeutics with the strongest correlations
3.6 CDCA8 was the hub gene in risk scoring modelsTo further evaluate the influence of the risk score model on the prognosis of HCC patients, this study first screened the model's hub gene CDCA8 using the STRING database and cytoscape software (Fig. 6A-B). The Sankey diagram results revealed a relationship between the expression of the hub gene CDCA8 and the age, grade, stage, and survival status of HCC patients, with patients with high CDCA8 expression clustering in the G3 and G4 grades, as well as the T3 and T4 stages (Fig. 6C). Survival analysis showed that patients with high CDCA8 expression had significantly lower OS (P = 4.06 × 10–5), PFS (3.85 × 10–5) and DFS (P = 0.0018) than HCC patients with low CDCA8 expression, which further demonstrated that high CDCA8 expression was closely associated with a poor prognosis in HCC patients (Fig. 6D-F). Functional analyses showed that the proliferative capacity of tumor cells (r = 0.879, P = 6.92 × 10–12) and EMT markers (r = 0.149, P = 4.05 × 10–3) increased with the increase in the expression of CDCA8, suggesting that CDAC8 may contribute to the development of HCC by promoting the proliferation and metastasis of tumor cells (Fig. 6G-H).
Fig. 6
Acquisition of the hub gene CDCA8 and its correlation with clinical features and pathologic factors. A and B Protein–protein interaction networks of risk genes; C sankey diagram of the distribution of survival status and clinicopathologic features of HCC patients between high and low expression groups of the hub gene CDCA8; D survival analysis of CDCA8 expression and OS in patients with HCC; E survival analysis of CDCA8 expression and PFS in patients with HCC; F survival analysis of CDCA8 expression and DFS in patients with HCC; G correlation analysis between CDCA8 expression and tumor cell proliferation; H correlation analysis between CDCA8 expression and EMT
3.7 CDCA8 may promote HCC progression by regulating T cellsThe correlation between the hub gene CDCA8 and the associated inflammatory response and anti-inflammatory function was further explored by functional analysis, which showed that the increased expression of CDCA8 did not correlate with the tumor inflammatory signature (P = 0.791), the inflammatory response (P = 0.294) and the IL-10 anti-inflammatory signaling pathway (P = 0.141) (Fig. 7A-C). Analysis of single-cell data showed that CDCA8 was mainly expressed in T lymphocytes and was concentrated in CD4+ T lymphocytes, CD8+ T lymphocytes and regulatory T cells, and the mean expression was highest in Tprolif cells (Fig. 7D-F).
Fig. 7
Correlation of the Hub gene CDCA8 with the inflammatory response and its expression in immune cells. A Correlation between CDCA8 expression and inflammatory signatures; B correlation of CDCA8 expression with inflammatory response; C correlation between CDCA8 expression and IL10 anti-inflammatory signalling pathway; D–F Analysis of the single-cell data showed that CDCA8 was expressed predominantly in T lymphocytes
留言 (0)