
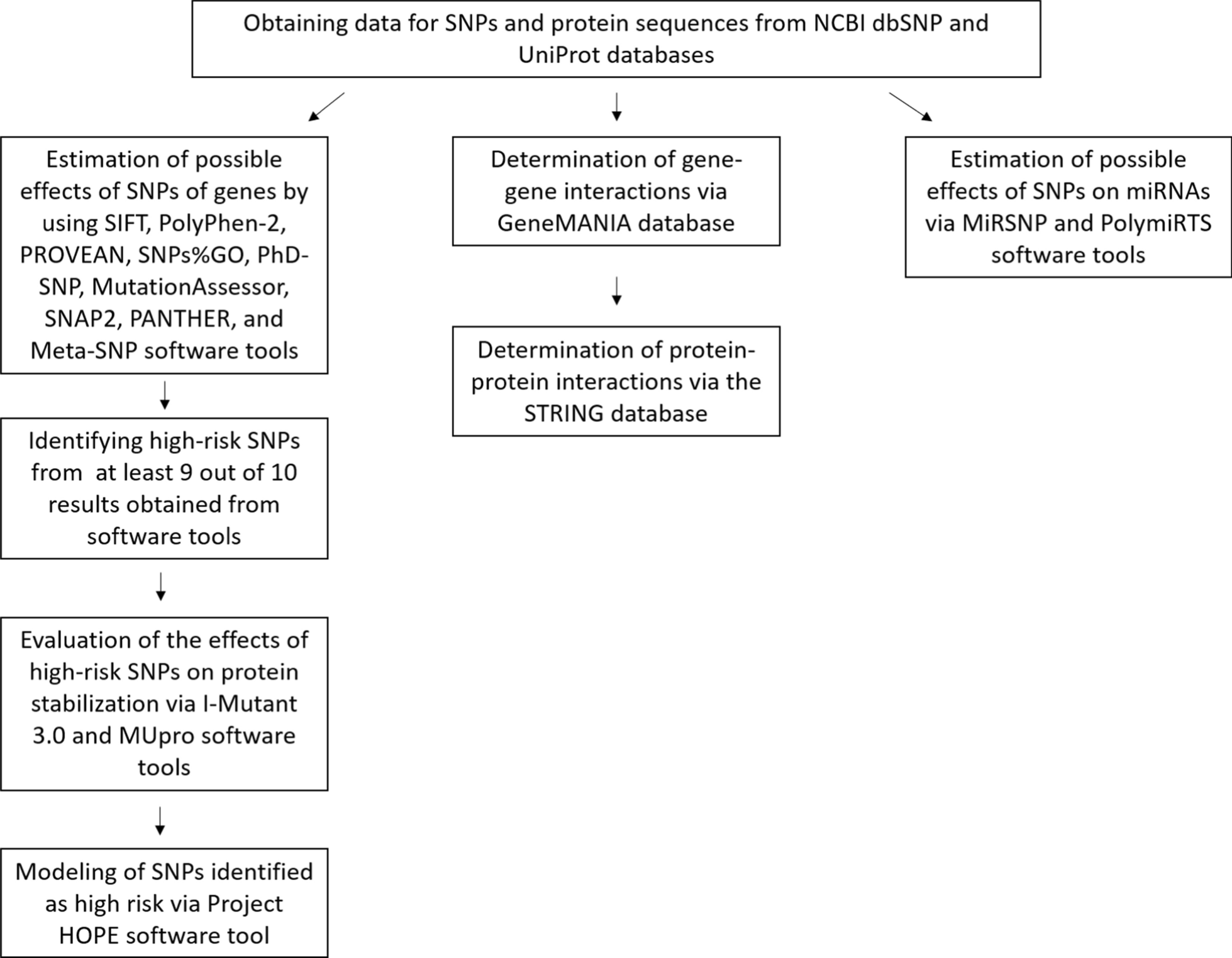
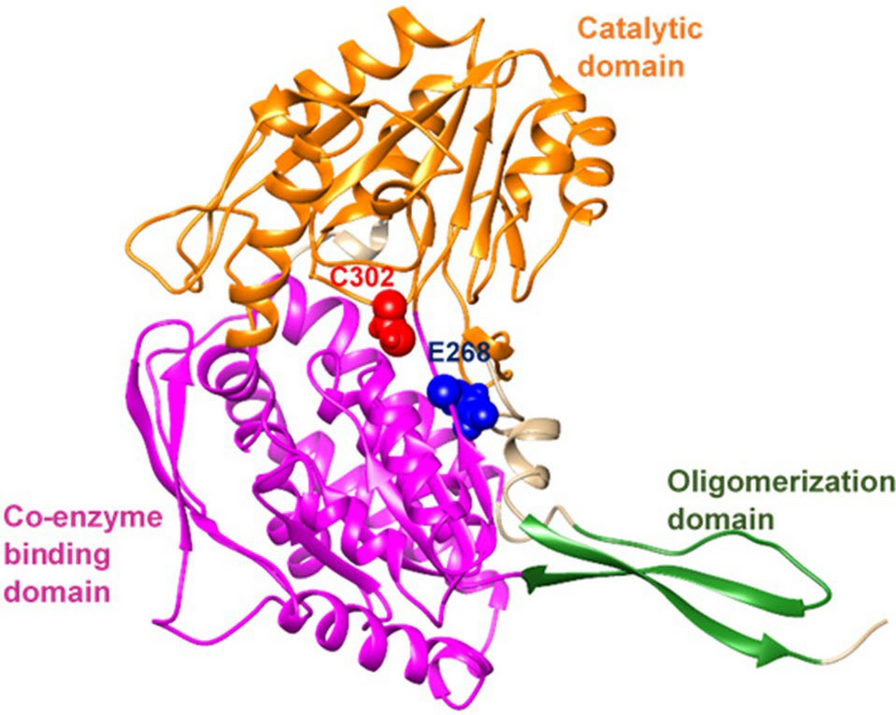
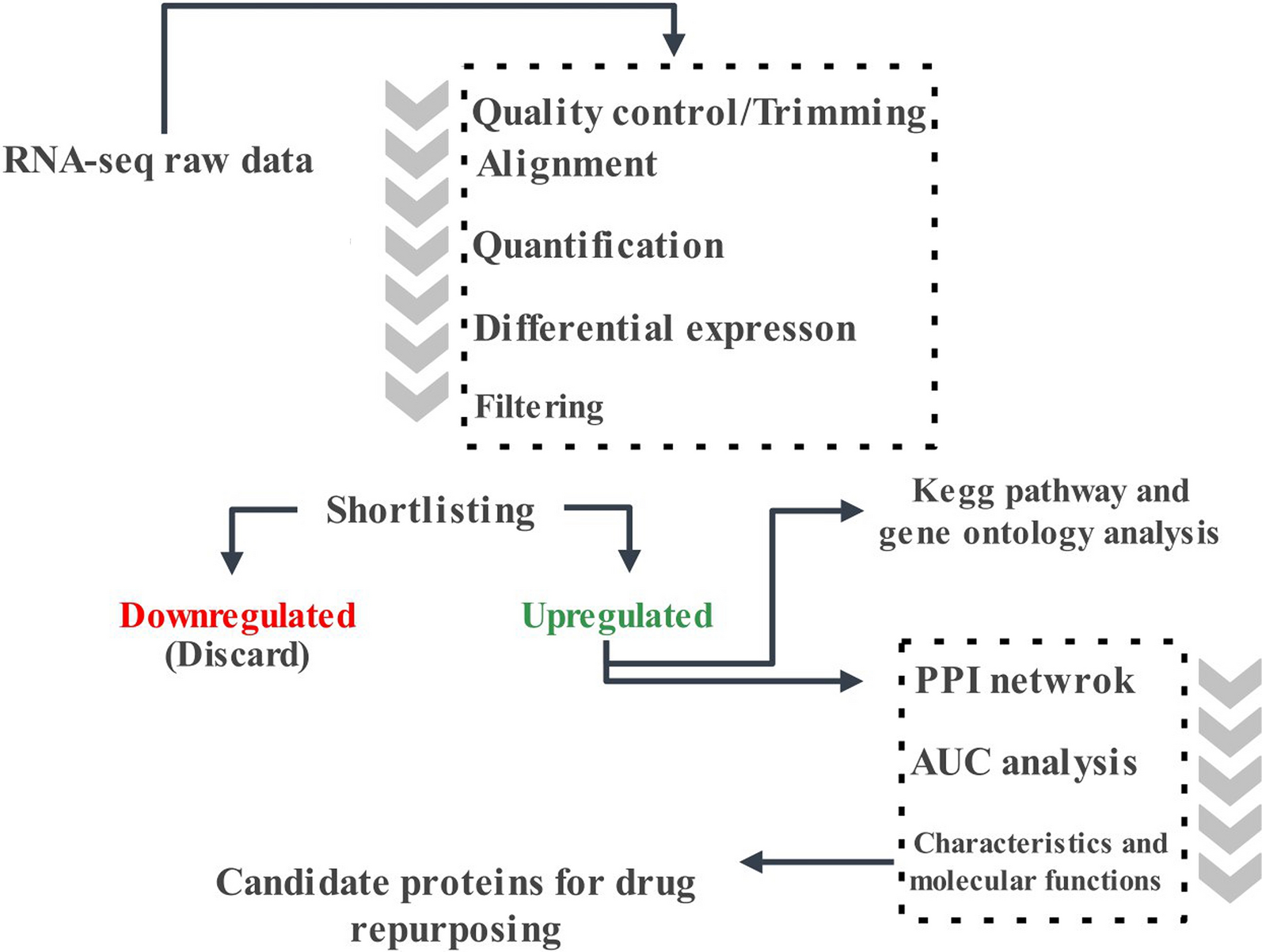
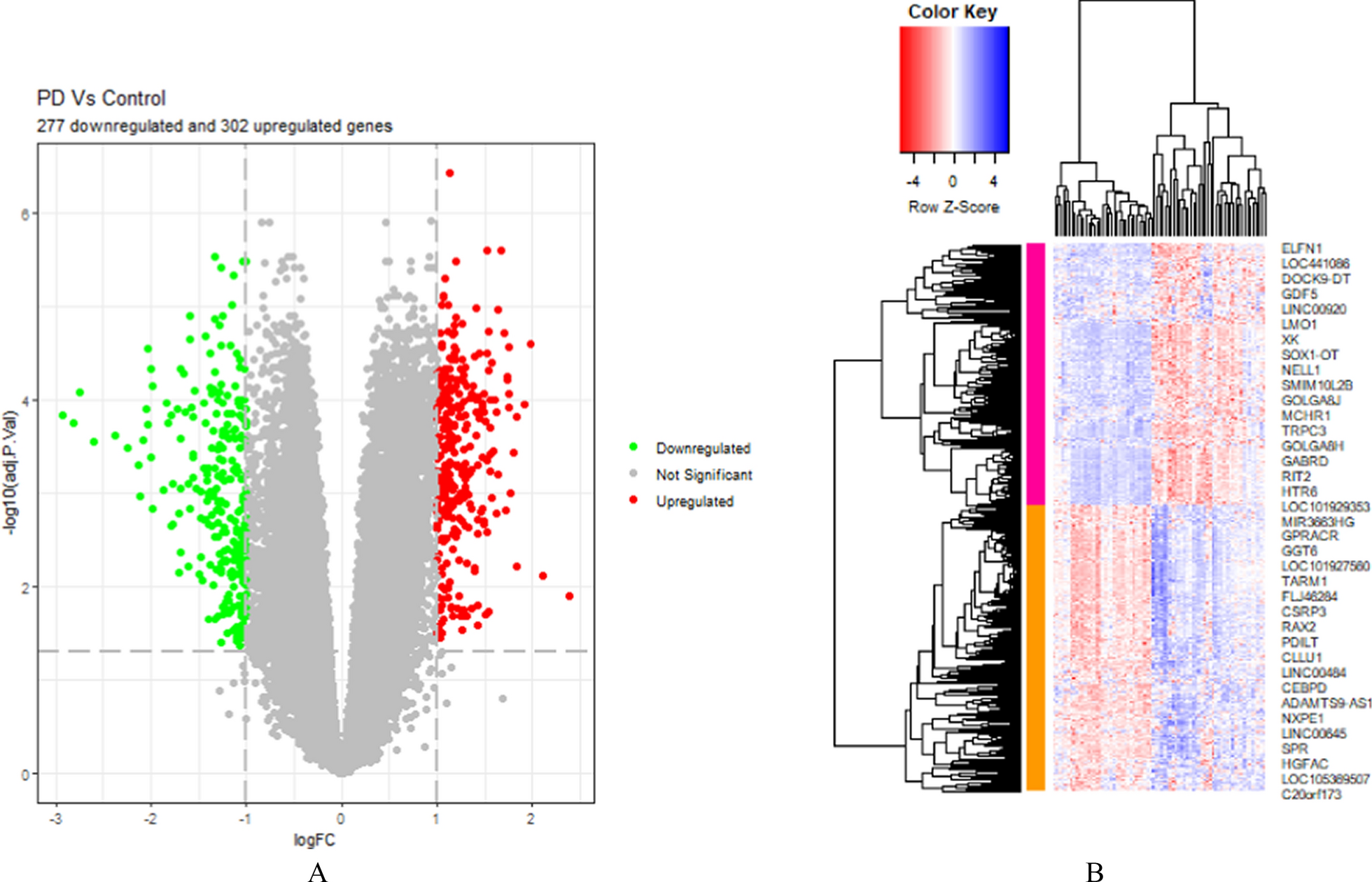
記住我





In general, ligand-induced oligomerization activates RTKs, resulting in phosphorylation of tyrosine residues in the activation and docking sites [4]. The phosphorylated tyrosine residues act as recruiting sites for the downstream effector proteins (Fig. 1). Autophosphorylation of RTKs activates several signaling pathways, including the MAPK, PI-3 K, and STAT pathways. Docking proteins act as assembly platforms, attracting more signaling molecules with SH2 or other domains and eventually activating transcriptional pathways that regulate growth, motility, and differentiation [12]. Different mechanisms can also activate receptor tyrosine kinases. In cancer, various pathways can mediate aberrant activation, including gain-of-function mutations, genomic amplification, chromosomal rearrangement, hypoxia-induced transcriptional activation, and autocrine activation (Fig. 2) [12].
Fig. 1
Mechanisms of aberrant activation of RTKs: overexpression, chromosomal translocation, gain-of-function mutations, and autocrine activation
Fig. 2
Receptors tyrosine kinases signaling pathways: when a ligand attaches to a tyrosine kinase receptor, it triggers the receptor's activation. This often leads to the formation of a dimer, where two receptor monomers pair up. The kinase domain within this receptor becomes active through phosphorylation of their kinase domains. Turning on the receptor, many downstream pathways are activated, including PI3K/AKT1, PLC/PKC, MAPK/RAS/ERK, SRC, and JAK/STAT signaling, resulting in increased proliferation, migration, survival, and cell cycle progression
C-MET is activated through interactions with a variety of membrane receptors, including EGFR, Integrin, b-catenin, CD44, ICAM-1, Plexin B1, VEGFA, INSR, FAS, MUC1, Nrp-1 and -2, and FAK. Independent of receptor activation, c-MET can signal under various cellular conditions, including apoptotic and necrotic stimuli, by generating c-MET receptor fragments [13]. C-MET overexpression, caused by hypoxia or mutations, can also drive receptor activation. Autocrine activation caused by ectopic MET expression in HGF-producing cells has previously been observed in acute myeloid leukemia [14]. The abnormal activation of HGF/c-Met signaling pathways can also be caused by gene amplification, ligand binding, or excessively high HGF levels, all of which contribute to tumor initiation and progression [15]. Amplification or upregulation of the HER2 gene may result in a higher HER2 protein on the tumor cell surface, producing an active heterodimer with the EGFR receptor and triggering signaling pathways that boost cell growth, proliferation, and survival [4]. P95, a mutant version of HER2 that lacks the extracellular domain and is constitutively active, was discovered in some breast cancer patients, contributing to resistance to HER2-targeted therapy [16]. Loss of PTEN function in BC may result in loss of the capacity to phosphorylate the Y1173 residue in HER2, leading to activation of the receptor. Furthermore, activating an alternate HER2 isoform lacking the phosphorylated Y1173 site may activate constitutive receptors [17]. HER2 activation can occur indirectly by activating cross-talking signaling pathways such as the PI3K/AKT pathway [18].
Clinic-pathological significance of c-MET and HER2 in bladder cancerTable 1 summarizes the findings of several studies that explored the involvement of c-MET and HER2 in BC patients. Early research found that c-MET was implicated in the development of BC in rat animal models even though the study relied on an established model, which may not capture the complexity of BC development in humans. In noncancerous cell lines, the c-MET pathway promoted cell proliferation and growth, whereas in malignant cell lines, it increased invasion and migration [19]. c-MET's capacity to induce migration and invasion in BC cells can be explained by modulating Akt/GSK-3β/snail signaling [20]. The c-MET, AXL, and PDGFR overexpression were reported in a previous study to enable the differentiation of BC cells from normal control tissues and predict disease progression in NMIBC patients. However, the study focused on the clinical relevance of c-MET network genes and a deeper exploration of molecular mechanisms is essential [21]. It was found that c-MET knockout reduced cell growth and increased apoptosis in vitro [21]. Another study investigated the expression of MET in bladder urothelium using (IHC) staining in 20 cases of bladder samples with inflammatory diseases and 142 BC patients. High MET expression was found in 4.9% of the patients, while 22.5% had low expression and 72.5% had none. Noncancerous tissues showed weak staining in the urothelium's basal cells [22]. It has been found that c-MET is expressed in a variety of urogenital malignancies, including BC, prostate cancer, and ovarian cancer. C-MET overexpression is ubiquitous in hyperactive tumors, exhibiting increased angiogenesis and neovascularization [15]. Such overexpression could occur because of transcriptional activation, hypoxia-induced overexpression, or MET amplification [23]. In prior research on the levels of c-MET and two phosphorylated tyrosine residues pY1349 and pY1234/pY1235, c-MET overexpression was related to muscle invasion (P = 0.021); pY1349 c-MET was significantly associated to the invasion of the muscle (P = 0.003), while pY1234/1235 c-MET was associated to both invasion (P = 0.006) and metastasis (P = 0.012), suggesting a prognostic and predictive role of c-MET in BC after exclusion of possible confounding factors including tumor grade, patient’s age, or treatment history [24]. The authors hypothesized that c-MET plays a significant role in muscle invasion by regulating PD-L1 and HO-1. In contrast, its phosphorylation at Y1349 is linked to muscle invasion and metastasis in BC patients via regulating COX-2, HO-1, and PD-L1 [24]. Furthermore, c-MET phosphorylation activates several intracellular signaling pathways, ultimately promoting motility, invasiveness, and epithelial–mesenchymal transition (EMT) [5]. Multiple variables contribute to carcinoma cells' invasive potential, including their capacity to undergo EMT [25]. In a previous study of 142 patients with BC, MET upregulation was significantly linked to nuclear grade (P < 0.05) [22]. C-MET expression was reported to be an independent predictor of lymph node metastasis in MIBC [26]. A review of BC recurrence and progression discovered that upregulation of c-MET was substantially linked to poor recurrence-free and progression-free survival rates. Inhibiting c-MET has demonstrated promise as a possible target for treating BC [6].
Table 1 Summary of studies investigating the role of c-MET and HER2 in BCHER2 expression was higher in BC, primarily due to gene amplification, and this phenomenon was linked to carcinogenesis [7]. HER2 overexpression in BC activates intracellular pathways that enhance cell proliferation, migration, and aggressiveness [31]. In addition, BC patients had a higher incidence of chromosome 17 polysomy and a higher HER2 copy number. Gene methylation can also influence HER expression, underscoring the importance of epigenetic regulation [32]. The level of HER2 expression in urothelial cancer ranged between 2 and 79% [33]. A recent study that examined HER2 protein by IHC in 50 BC specimens found that HER2 expression was directly associated with tumor grade and stage; long-term follow-up is needed to establish a definitive relation with the BC patient’s outcome [27]. Increased HER2 gene expression is associated with BC lymph node invasion. A prior meta-analysis found that HER2 protein was substantially elevated in multifocal cancers [31]. A retrospective analysis of 40 paraffin blocks recovered from BC cases by IHC discovered that HER2-positive expression was identified in 25% of NMIBC cases and 90% of MIBC cases, giving into consideration limitations of IHC, including subjectivity in scoring, antibody specificity, and sensitivity [30]. A recent report pointed to the possible participation of HER2 in EMT in some malignancies, boosting their invasive capacities via cadherin-driven EMT signaling pathways within tumor cells [30]. Patients with high HER2 protein expression showed a higher risk of tumor recurrence and progression compared to those with low expression [11].
Crosstalk between c-MET and HER2 in cancerIncreasing data suggests that abnormally activated RTKs may not operate in isolation but rather interact with other pathways in healthy and altered cells, a phenomenon known as "RTK co-activation." RTK co-activation can promote tumor growth via numerous methods, as shown in Fig. 3. Nowadays, viewing RTKs as isolated entities is insufficient; instead, these receptors should be studied as part of networks or receptor partners [12].
Fig. 3
Receptor tyrosine kinases co-activation mechanisms: co-activation of RTKs can enhance tumor growth, survival, and resistance to cancer treatments. A RTKs can activate a shared repository of SH2 and adaptor proteins containing PTB, allowing subsequent oncogenic signaling to proceed. B Single RTKs can regulate a variety of survival pathways. When one receptor is blocked, subsequent effector signaling decreases; other RTKs' effectors vigorously promote alternate survival pathways. C Given that heterogeneous signaling from specific RTKs can contribute to various cancer phenotypes, RTK co-activation can increase the variety of malignant features, such as invasion and potential for metastatic spread
Many examples of such "crosstalk" have been identified involving the HGF/c-MET axis, HER2, and other RTKs, which may allow for the emergence of tumor cell resistance to targeted inhibition of this pathway in isolation. The co-expression status of c-MET, FGFR2, and HER2 in gastric cancer (GC) and their clinical implications in therapy were previously explored. In the RTK's co-expression study, 3.5% of the patients were triple positive. Patients with triple-positive GC had the lowest overall survival [34].
Co-expression of MET and HER2 was identified in breast cancer, which contributed to RTK inhibitor resistance caused by MET amplification or autocrine synthesis of MET ligands. Furthermore, a prior study found that a considerable number of breast cancer tumors co-expressed MET and HER2 receptors and that MET depletion enhanced HER2 activation while HER2 depletion increased MET activation. Furthermore, examining RTK activation during HER2 knockdown suggested that increased MET signaling could serve as a compensatory resistance pathway [35]. In esophagogastric cancer, c-MET amplification caused resistance to afatinib, an anti-HER2 antibody used as a standard agent for managing advanced esophagogastric cancer with HER2 overexpression. The combination of afatinib and a MET inhibitor was advised [36]. Similarly, MET and HER2 overexpression in lung cancer plays a crucial role in developing radio- and chemoresistance by suppressing apoptosis via activation of the PI3K-AKT pathway [37]. The possible interplay between c-MET and HER2 in BC wasn’t fully elucidated; further research exploring c-MET and HER2 signaling axis is warranted and could offer valuable insights into identifying novel therapeutic targets.
Targeting c-MET, HER2 in BC, and further directionsThere has been much interest in targeting the RTK pathway in BC, with multiple clinical trials looking into drugs that target c-MET and HER2. C-MET interacts with other signaling molecules (such as EGFR, VEGFR, and RON) activating pathways that mediate tumor progression and drug resistance. Cabozantinib, a nonselective tyrosine kinase inhibitor (TKI), targets c-MET and VEGFR. It is used as a second-line treatment for metastatic BC. A phase II clinical trial that combined cabozantinib with platinum-based chemotherapy indicated a 43.8% response rate [38]. A preclinical study demonstrated that crizotinib (another TKI targeting c-MET) and cabozantinib significantly suppressed the HGF/c-MET-induced tumor cell growth and invasion in the BC cell lines [39]. Monoclonal antibodies, like onartuzumab and emibetuzumab, which target c-MET, have been used to treat gastric tumors but not BC patients. Chimeric antigen receptor (CAR) T cell therapy, which involves genetically modified T cells targeting specific antigens, has shown promise in mouse models of papillary renal cell carcinoma [38].
In several prospective therapeutic trials, HER2 was targeted in monotherapy, in combination with other anti-HER2 agents, or in other systemic treatments. Lapatinib has been evaluated in several clinical trials in combination with platinum chemotherapies and has shown disappointing results. Afatinib, on the other hand, showed better results, according to three phase II clinical trials [40]. Ongoing phase II and III clinical studies using antibody–drug conjugates (ADCs) targeting HER2 have also yielded positive findings. It has been proposed that HER2 and c-MET signaling can increase PD-L1 expression. Preliminary evidence suggests that targeting both PD-(L)1 and HER2 or c-MET has synergistic effects [41].
Overall, targeting c-MET and HER2 provides a potential approach to treating BC, and more studies are needed to determine the appropriate usage of these medicines in clinical practice. Given the complex nature of BC, combination strategies targeting multiple signaling pathways hold the potential for enhancing therapeutic efficacy and overcoming resistance. Crosstalk between c-MET and HER2 was evident in cancer, so further trials investigating the use of novel agents that target c-MET together with HER2 in BC are recommended, either as monotherapy or in combination with other treatments.
留言 (0)