
記住我
Critical illness, marked by organ-level pathophysiological disruptions, remains a significant global health challenge (1, 2). Despite its impact, intensive care unit (ICU) outcomes are often underrepresented in global disease burden studies. For instance, a 2017 report revealed that sepsis-related deaths accounted for 47% of all fatalities worldwide, underscoring an urgent need for focused research (2, 3). While advancements in critical care have improved symptomatic management—such as organ support and fluid resuscitation—progress in targeted therapies has been limited. Identifying novel mechanisms and therapeutic strategies is, therefore, paramount. Mechanisms linked to metabolic dysregulation, including ferroptosis, autophagy, and oxidative stress—processes driven by lipid peroxidation and cellular ion imbalances—present promising avenues for innovation in critical care.
Cell death is a fundamental process critical for growth, homeostasis, and the progression of various diseases (2, 4). Programmed cell death pathways, such as necrosis, autophagy, pyroptosis, and apoptosis, operate through well-defined signaling regulatory mechanisms and are closely tied to disease pathophysiology (2, 5, 6). Among these, ferroptosis—an iron-dependent form of regulated cell death triggered by lipid peroxidation—has emerged as a key contributor to critical illnesses, including sepsis, acute respiratory distress syndrome (ARDS), acute kidney injury (AKI), and Ischemia-reperfusion injuries (IRI) (2, 7–9). Growing evidence underscores the importance of targeting ferroptosis to better understand and manage these life-threatening conditions. Iron, an essential trace element in the human body, is involved in numerous biological processes, including energy metabolism and nucleotide synthesis and repair (2, 10). While the concept of ferroptosis dates back to the 1980s, it was formally named by Dixon in 2012 (2, 7, 11). Ferroptosis development is driven by iron-induced reactive oxygen species (ROS), making it susceptible to inhibition by lipophilic antioxidants and agents such as ferritin and iron chelators (2, 12).
Current evidence highlights ferroptosis as a key player in the onset and progression of various diseases, positioning it as a potential target for clinical therapies (Figure 1). This review summarizes the occurrence, characteristics, regulatory mechanisms, and critical molecular pathways of ferroptosis, emphasizing its direct and indirect roles in the etiology of serious disorders. Additionally, we explore potential therapeutic targets linked to ferroptosis, offering new insights for clinical applications in treating critical conditions.
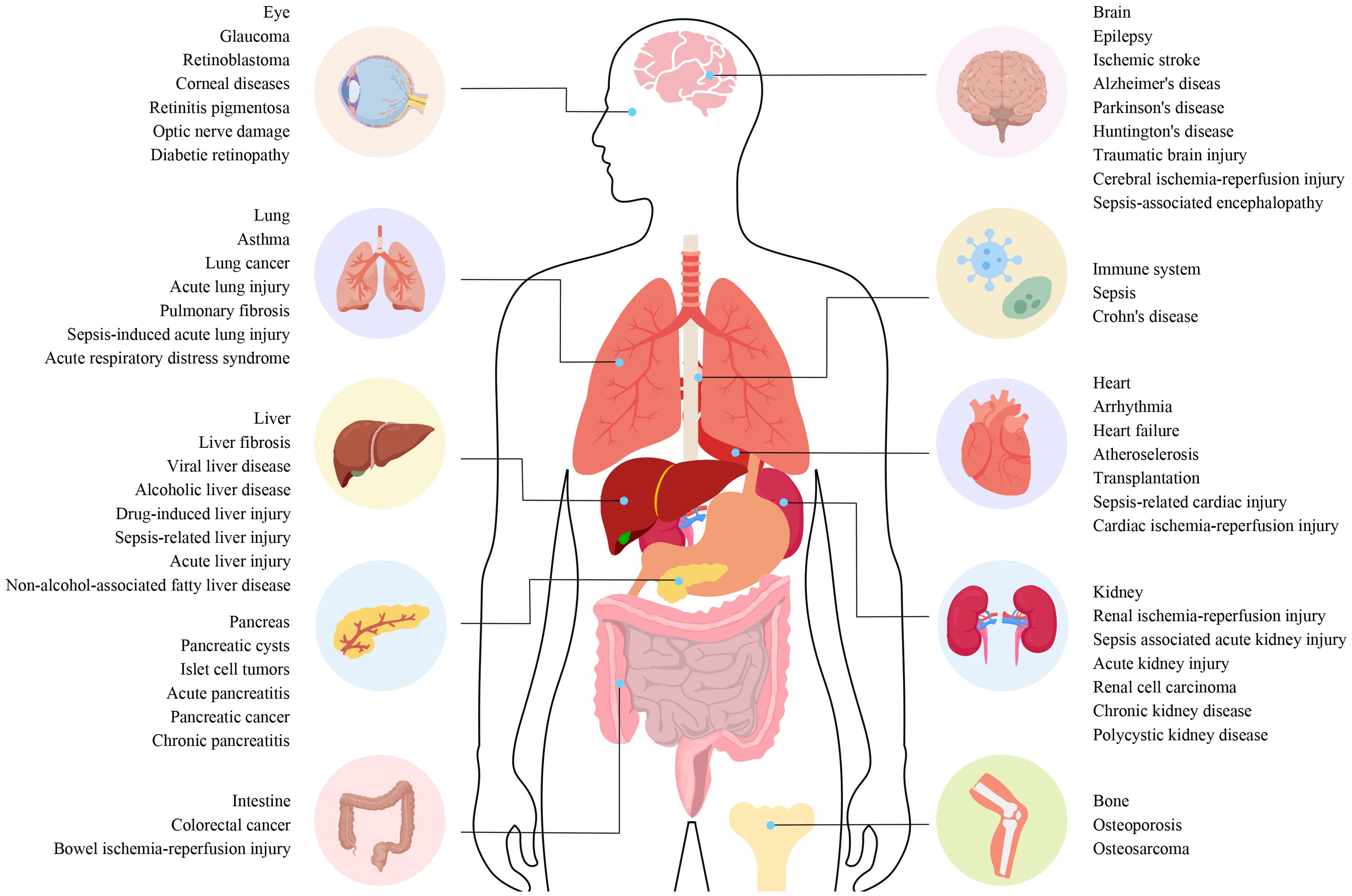
Figure 1. The connection between different diseases and ferroptosis. The connection between different diseases and ferroptosis. Ferroptosis plays a role in the regulation of various systemic diseases, such as diseases of the nervous system, cardiovascular system, digestive system, musculoskeletal system, autoimmune system, visual system, lung, liver, and kidney. Ferroptosis often involves systemic interactions, where iron metabolism and oxidative stress affect multiple organ systems. For example, diseases like sepsis can trigger widespread inflammation and organ failure involving ferroptotic pathways in the liver, lungs, kidneys, and cardiovascular system. Understanding these pathways is essential for developing targeted therapies that can mitigate ferroptosis-induced damage across various diseases.
2 Discovery of ferroptosisIn 1980, System Xc⁻ was identified as a transporter that exchanges glutamate for cystine, enabling cystine entry into cells (2, 13). In 2003, Dolma et al. discovered that NSC146109 (later known as erastin) selectively killed BJeLR cells with mutated Ras oncogenes during high-throughput screening (2, 14). In 2008, compounds like RAS-selective lethal small molecules 3 (RSL3) and RSL5 were identified as non-apoptotic agents that induce cell death (2, 15). This form of cell death is distinct from previously recognized categories, as apoptosis, necrosis, and autophagy inhibitors did not prevent cell death induced by these compounds. However, significant inhibition could be achieved using iron chelators and lipid peroxide inhibitors (2, 16). In 2012, Dixon et al. demonstrated that erastin, a product linked to the Ras oncogene, induced cell death in tumor cells and coined the term ferroptosis for this iron-dependent mode of cell death (2, 11). The concept of ferroptosis gained formal recognition in 2018 when the Cell Death Nomenclature Committee defined it as a potentially regulatory mode of cell death (2, 17).
3 Ferroptosis and other types of cell deathFerroptosis differs from other forms of programmed cell death by exhibiting distinct morphological features (2, 18). These include a reduction or absence of mitochondrial cristae, rupture of the outer mitochondrial membrane, mitochondrial shrinkage, and an increase in membrane density. While nuclear size remains normal, chromatin condensation is absent, and membrane blebbing occurs without full rupture (2, 19, 20). Biochemically, ferroptosis is marked by elevated iron and ROS levels, decreased glutathione (GSH), and reduced glutathione peroxidase 4 (GPX4) activity. It also involves disruption of the cysteine uptake system, changes in mitochondrial membrane potential, and arachidonic acid-mediated release of functional factors, all leading to lipid peroxidation and mitochondrial dysfunction. Additionally, specific gene expression changes have been observed, and ongoing studies are exploring the relationship between ferroptosis and other forms of cell death (21).
3.1 Ferroptosis and apoptosisBoth ferroptosis and apoptosis are programmed cell death mechanisms, but they differ in their biological characteristics, triggers, and signaling pathways, though there are notable intersections (22). For instance, B-cell lymphoma-2 (Bcl-2) family proteins, key regulators in the mitochondrial pathway of apoptosis, also influence ferroptosis. These proteins interact with Beclin1 (BECN1) to inhibit autophagy and ferroptosis, prevent cytochrome c release from mitochondria in apoptosis to block cell death, and regulate lipid metabolism and redox status in ferroptosis to prevent its occurrence (23, 24). Additionally, in cancer therapy, inducing ferroptosis is considered a strategy to target apoptotic cancer cells, particularly those resistant to conventional apoptosis-inducing treatments.
3.2 Ferroptosis and necroptosisIn some cases, the signaling pathways of ferroptosis and necroptosis converge. For example, under certain stress conditions, receptor-interacting protein kinase 1 (RIPK1) not only mediates necroptosis but can also influence ferroptosis by modulating cellular redox status (25). RIPK1 regulates the activity of antioxidant enzymes, indirectly affecting lipid peroxide levels and either promoting or inhibiting ferroptosis (26). During ferroptosis, cell membrane rupture releases several damage-associated molecular patterns (DAMPs), such as high-mobility group box 1 (HMGB1), which activate immune cells and trigger inflammation. Necroptosis, an inflammatory mode of cell death, similarly leads to membrane disruption through mixed lineage kinase domain-like protein (MLKL), causing the release of numerous inflammatory factors (27). Inflammation-related diseases may involve both ferroptosis and necroptosis, acting synergistically to exacerbate tissue damage and inflammation.
3.3 Ferroptosis and necrotic deathThe transition from necrotic death to ferroptosis can be facilitated by phosphatidylethanolamine binding protein 1 (PEBP-1) and 15-lipoxygenase (28). Belavgeni et al. suggest that necrotic death may represent an initial phase in the ferroptosis-mediated spread of cell death (29). A small-molecule inhibitor, necrostatin-1f, was developed to investigate this relationship. Although it effectively inhibits necrotic death, it weakly inhibits ferroptosis, highlighting the distinct yet interconnected nature of these cell death pathways (30).
3.4 Ferroptosis and pyroptosisPyroptosis can be induced by ROS-generating drugs and iron ions through the ROS-Tom20-Caspase3-GSDME signaling pathway (31). During ferroptosis, lipid peroxides break down into 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA), which interact with proteins, nucleophiles, and DNA bases, causing significant cytotoxicity (32). This process amplifies ROS signaling, activating mitochondrial caspase pathways typically associated with pyroptosis. These findings suggest a potential link between ferroptosis and pyroptosis, indicating an overlap between these cell death mechanisms.
3.5 Ferroptosis and autophagyFerroptosis regulation and progression are tightly linked to selective autophagy, which facilitates the degradation of key ferroptosis-related molecules and organelles. Recent studies highlight autophagy’s role in controlling ferritin levels, the primary iron storage protein (7). Nuclear receptor coactivator 4 (NCOA4), a transport receptor that selectively degrades ferritin, is a key player in regulating intracellular iron homeostasis. Overexpression of NCOA4 enhances ferritin degradation, increasing labile iron levels, which promotes ferroptosis (33).
3.6 Ferroptosis and oxidative deathFirst described in 2001, oxidative death is a non-apoptotic form of cell death characterized by glutathione depletion and oxidative stress (34). This process shares similarities with ferroptosis, as both activate the expression of eIF2α (35). Moreover, studies show that CRISPR/Cas9-mediated gene knockdown can protect synapses from both ferroptosis and oxidative death (36).
3.7 Ferroptosis and cuproptosisCuproptosis, a recently identified form of metal ion-dependent cell death, is driven by intracellular copper levels. Excess copper directly binds to fatty acylated components of the tricarboxylic acid cycle (TCA), disrupting protein homeostasis and causing the accumulation of fatty acylation-related proteins, ultimately triggering cell death (37). Recent studies suggest that ferroptosis inducers can initiate and accelerate cuproptosis in liver cancer. The concurrent use of ferroptosis and cuproptosis inducers leads to enhanced cell death (38).
3.8 Ferroptosis and disulfidptosisCancer cells with high levels of solute carrier family 7 member 11 (SLC7A11) undergo disulfidptosis, a thiol-dependent cell death induced by disulfide stress. This process is marked by the formation of multiple disulfide bonds in the cytoplasm, particularly under glucose starvation. This emerging phenomenon suggests a potential link between disulfidptosis and ferroptosis (39). However, evidence on the interaction between these two processes remains limited, and future research is needed to explore and clarify their relationship.
4 Regulatory mechanisms of ferroptosis4.1 Regulation of iron homeostasisIron is essential for various physiological functions, and its homeostasis is crucial to preventing ferroptosis (40). Iron metabolism, which includes absorption, activation, storage, and recycling, is tightly regulated to ensure cellular health. Iron primarily enters the bloodstream through dietary intake and macrophage-mediated erythrocyte breakdown. It is absorbed in the Fe3+ form, binds to transferrin (TF), and is recognized by transferrin receptor 1 (TfR1) on cell membranes, after which the complex is internalized through endocytosis (41). TfR1 has been suggested as a potential marker for ferroptosis (42).
Inside cells, ferric reductase six-transmembrane epithelial antigen of prostate 3 (STEAP3) reduces Fe3+ to Fe2+, which binds to ferritin or is transported by ferroportin (Fpn) out of the cell. Divalent metal transporter 1 (DMT1) imports free Fe2+ from nucleosomes into the cytoplasm, contributing to the labile iron pool (LIP), where it plays a role in metabolic processes (43–45). Ferroportin and enzymes such as ceruloplasmin (CP) export Fe2+ from cells, converting it back to Fe3+ to maintain the iron cycle (46).
Iron homeostasis is tightly regulated by iron response elements (IREs) and iron regulatory proteins (IRPs), which control the translation of genes such as DMT1 and TfR1. Iron-regulated protein 2 (IRP2) binds to IREs under low-iron conditions to regulate iron levels, while IRP1 acts as a coenzyme when bound to an Fe-S cluster. When iron levels are high, IRP2 is degraded, and IRP1 remains bound to the Fe-S cluster, inhibiting further regulation and stabilizing iron homeostasis (47, 48).
The hepcidin-ferroportin-1 (Fpn1) axis is pivotal for iron regulation (49). Ferroportin exports iron, while ferritin stores excess iron (41, 50). In ferroportin-deficient mice, iron homeostasis is severely disrupted, leading to iron overload (51). Excessive iron, primarily stored in ferritin or as free Fe2+, can induce ferroptosis. Iron can overwhelm TF binding capacity, resulting in non-transferrin bound iron (NTBI), which catalyzes harmful reactions, such as Fenton and Haber-Weiss, producing ROS (52). These ROS damage lipids, driving ferroptosis. The transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2), a leucine zipper protein, plays a crucial role in reducing excess iron and preventing ferroptosis by regulating iron intake, storage, and recycling.
In IRI, tissue damage occurs due to hypoxia during ischemia, and subsequent reperfusion further exacerbates ROS generation. Iron participates in the Fenton reaction, catalyzing the conversion of hydrogen peroxide into free radicals, thus intensifying cellular oxidative damage and potentially triggering ferroptosis. In sepsis, the inflammatory response significantly alters iron metabolism, redistributing iron from storage sites and decreasing serum iron levels. Excessive iron accumulation in cells and the reticuloendothelial system promotes ROS generation, induces ferroptosis, and exacerbates organ dysfunction. Inflammatory mediators in septic patients also regulate iron metabolism-related proteins, such as increasing hepcidin expression to drive iron accumulation. This retention and overload activate oxidative stress responses, damaging cellular structures and leading to tissue damage and organ failure.
4.2 Lipid metabolismFerroptosis is strongly influenced by lipid peroxidation, especially through polyunsaturated fatty acids (PUFAs), which undergo peroxidation in the presence of lipoxygenases and ROS. As integral components of cell membranes, PUFAs play key roles in immune response, inflammation, and cell proliferation (50). Free PUFAs, acting as precursors for lipid signals, must first esterify into membrane phospholipids and undergo oxidation to transmit ferroptosis signals. This process is facilitated by lysophosphatidylcholine acyltransferase 3 (LPCAT3) and acyl-CoA synthetase long-chain family member 4 (ACSL4) (53, 54).
Adrenoic acid (AdA) and arachidonic acid (AA) are primary PUFAs involved in inducing ferroptosis (54). ACSL4, in association with Coenzyme A (CoA), forms AA-CoA or AdA-CoA intermediates, which LPCAT3 esterifies into phosphatidylethanolamines, such as PE-AA or PE-AdA. These phospholipids are oxidized by lipid oxidases (LOXs) or via autoxidation, leading to cell death (53). ACSL4 also upregulates ferroptosis in a feedback loop, particularly when the neurofibromin 2-yes-associated protein 1 (NF2-YAP) pathway is inhibited (55).
Phosphorylation of ACSL4 enhances lipid peroxidation and facilitates the incorporation of PUFAs into plasmalogens, a process mediated by protein kinase C beta type isoform 2 (PKCβII) (55). In contrast, integrin α6β4 inhibits ferroptosis by downregulating ACSL4 expression. Moreover, thiazolidinedione hypoglycemic agents and safranin effectively suppress ferroptosis through ACSL4 targeting (56, 57). Knockdown of LPCAT3 confers protection against ferroptosis, underscoring its critical role (54, 57).
LOXs catalyze the oxidation of PUFAs both directly and within biofilms (53, 58). Among them, 15-LOX is particularly involved in lipid peroxidation, with its interaction with PUFAs, such as sn2-15-hydroperoxy-eicosatetraenoyl-phosphatidylethanolamines (sn2-15-HpETE-PE), playing a pivotal role in signaling ferroptosis (59). The involvement of 12-LOX in p53-mediated ferroptosis remains debated, despite its acknowledged importance (60).
Lipophagy, the autophagic degradation of lipid droplets (LDs), regulates ferroptosis by modulating lipid peroxidation (61). Inhibiting RAB7A or promoting tumor protein D52 (TPD52)-mediated lipid storage can prevent ferroptosis (61, 62). A deeper understanding of lipid peroxidation and its regulatory enzymes opens new therapeutic avenues for diseases such as cancer.
During the ischemic phase of IRI, oxygen and nutrient supply to cells is insufficient, disrupting energy metabolism and lipid homeostasis. Reperfusion leads to the influx of oxygen and iron ions, triggering oxidative reactions. Unsaturated fatty acids, particularly those in phospholipids containing polyunsaturated fatty acids, are prone to oxidation and serve as primary substrates for lipid peroxidation (62). Additionally, IRI affects the expression and activity of enzymes involved in lipid metabolism, altering intracellular lipid composition and further disrupting cellular homeostasis.
Sepsis accelerates the mobilization and oxidation of fatty acids, leading to an accumulation of lipid peroxidation byproducts such as 4-HNE and MDA. These products are toxic to cells, compromising membrane integrity and inducing cell death through ferroptosis (63). The inflammatory response during sepsis promotes fatty acid mobilization and increases the proportion of polyunsaturated fatty acids in cell membranes, heightening susceptibility to lipid peroxidation. This is particularly detrimental in endothelial cells and leukocytes, where lipid peroxidation and iron overload synergistically exacerbate cell damage and contribute to multi-organ failure. Sepsis-induced inflammation also boosts ROS production, further enhancing lipid peroxidation and establishing a vicious cycle that amplifies cellular damage.
4.3 Mitochondrial dysfunctionMitochondria, essential for lipid and energy metabolism in eukaryotic cells, are pivotal in regulating ferroptosis (64, 65). They synthesize heme and Fe-S clusters, which are critical for ferroptosis regulation (66, 67). Iron enters mitochondria through mitoferrin 1 (SLC25A37) in erythrocytes and mitoferrin 2 (SLC25A28) in non-erythrocytes (68). Overexpression of SLC25A28 can lead to redox-active iron accumulation, increasing ferroptosis susceptibility (69). Conversely, reducing SLC25A28 activity can prevent erastin-induced ferroptosis.
Heme acts as a cofactor in metabolism and electron transfer (70). Excess heme is either exported by feline leukemia virus subtype C receptor-related protein 1B (FLVCR1B) or metabolized by heme oxygenase 1 (HO-1) into Fe2+, carbon monoxide (CO), and biliverdin (71). While HO-1 activation can induce ferroptosis by causing iron overload, modest upregulation of HO-1 confers protection against ferroptosis (72, 73).
Cysteine desulfurase (NFS1) is essential for Fe-S cluster synthesis, and its inhibition sensitizes cancer cells to ferroptosis (74). Increased expression of mitoNEET (CISD1) inhibits erastin-induced ferroptosis, while overproduction of NAF1 (CISD2) prevents sulfadiazine-induced ferroptosis (75, 76). The ABCB7 transporter, critical for mitochondrial iron homeostasis, also plays a role in ferroptosis regulation (77).
Overexpression of mitochondrial ferritin (FtMt) reduces erastin-induced ferroptosis by enhancing iron storage and lowering the LIP, indicating a protective role (78, 79). Voltage-dependent anion channels (VDACs), located in the outer mitochondrial membrane, facilitate chemical and energy transport and are involved in programmed cell death (80). Yagoda et al. demonstrated that knockdown of VDAC2/3 reduces erastin sensitivity in tumor cells (81). Erastin targets VDAC2/3, causing mitochondrial dysfunction and increased ROS production, suggesting that mitochondria are potential therapeutic targets for mitigating ferroptosis.
In IRI, the ischemic phase disrupts mitochondrial energy supply, decreases adenosine triphosphate (ATP) production, and causes metabolic disturbances. Although oxygen re-supply during reperfusion restores mitochondrial activity, it also dramatically increases ROS generation due to the sudden activation of the electron transport chain (82). Iron overload within mitochondria exacerbates ROS accumulation and lipid peroxidation, triggering ferroptosis. Additionally, mitochondrial membrane damage releases signaling molecules that activate downstream cell death pathways, further exacerbating tissue damage.
Sepsis, a complex systemic inflammatory response, often results in severe mitochondrial dysfunction. The release of inflammatory mediators, oxidative stress, and increased metabolic load contribute to mitochondrial damage. Disrupted iron metabolism and mitochondrial iron overload are particularly prominent in sepsis, making mitochondria a major source of ROS (83). Overproduction of mitochondrial ROS worsens lipid peroxidation, triggering ferroptosis and promoting organ dysfunction and failure. Furthermore, mitochondrial dysfunction in sepsis impairs antioxidant defenses, notably by reducing GPX4 activity. This impairs lipid peroxide reduction, fostering ferroptosis. Loss of mitochondrial function also disrupts cellular energy metabolism, diminishes the ability to manage oxidative and metabolic stress, and increases susceptibility to ferroptosis.
4.4 System Xc⁻-GSH-GPX4 axisThe System Xc⁻–GSH–GPX4 axis, a crucial component of the antioxidant system, plays a pivotal role in inhibiting ferroptosis. System Xc⁻ facilitates cystine uptake, enabling GSH synthesis, which, in turn, supports GPX4 in reducing peroxides, maintaining cellular stability, and preventing ferroptosis (84). Dysregulation of this axis influences ferroptosis and contributes to various pathologies (Figure 2).
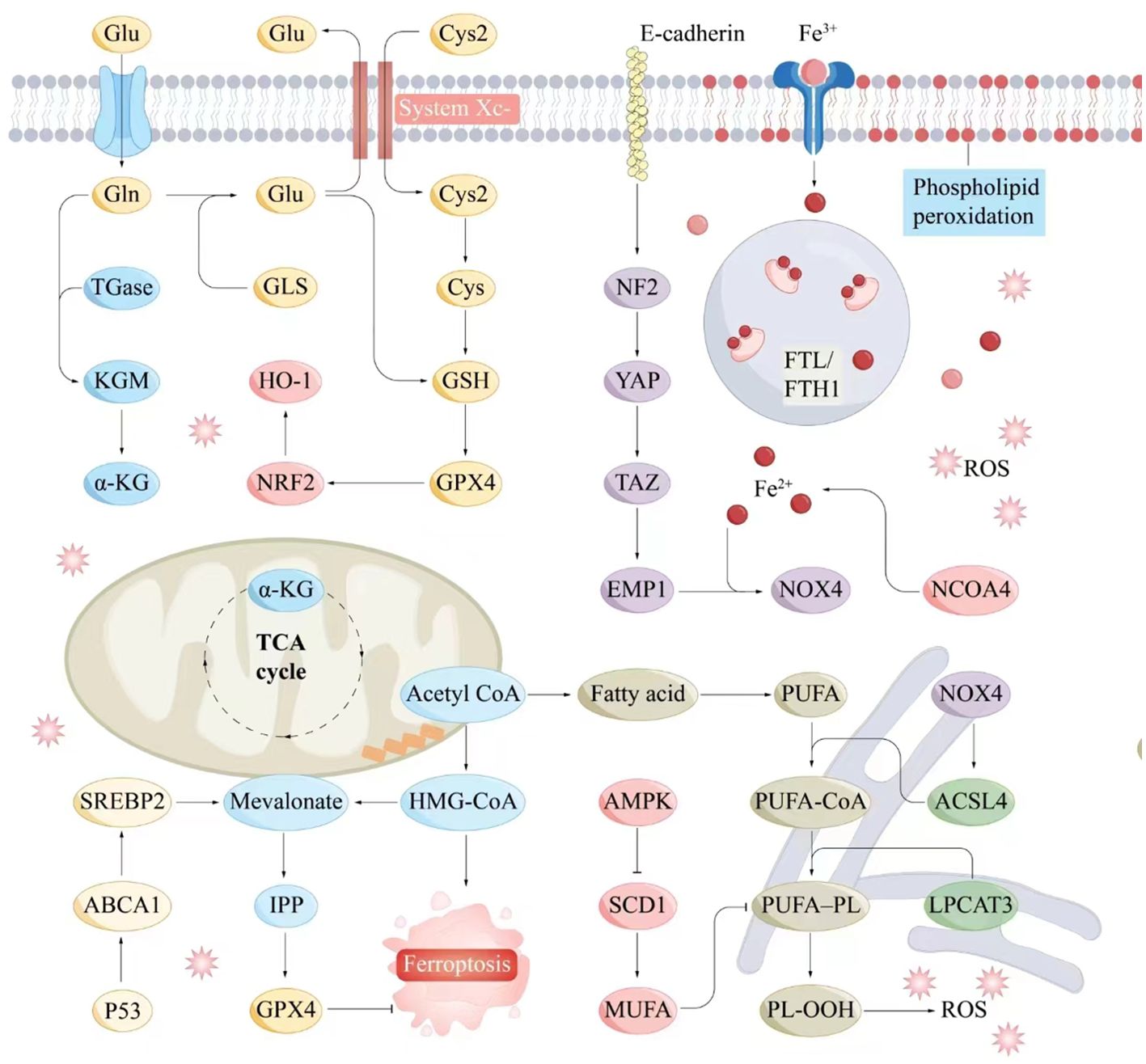
Figure 2. Molecular interaction diagram related to ferroptosis mechanisms. Key molecules and pathways involved in the process of ferroptosis include iron metabolism, antioxidant responses, lipid peroxidation, and related signaling pathways. Iron metabolism plays a central role in ferroptosis, as the accumulation of iron within cells can catalyze the formation of ROS through the Fenton reaction. Antioxidant Responses include GSH, GPX4, NADPH and FAD. Serve as electron donors in the regeneration of GSH and other antioxidants, supporting the cellular antioxidant response to counteract ferroptosis. Lipid peroxidation is a hallmark of ferroptosis, involving the oxidative degradation of PUFAs in cellular membranes. ABCA1, ATP-binding cassette transporter A1; Acetyl CoA, Acetyl coenzyme A; ACSL4, Acyl-CoA synthetase long-chain family member 4; AMPK, AMP-activated protein kinase; α-KG, Alpha-Ketoglutarate; Cys, Cysteine; E-cadherin, Epithelial cadherin; EMP1, Epithelial membrane protein-1; Fe3+, Iron ion (trivalent); Fe2+, Iron ion (divalent); FTL/KGM, Ferritin light chain/keratinocyte growth factor; FTH1, Ferritin heavy chain 1; Gln, Glutamine; Glu, Glutamate; GLS, Glutaminase; GPX4, Glutathione peroxidase 4; GSH, Glutathione; HMG-CoA, 3-Hydroxy-3-methylglutaryl coenzyme A; HO-1, Heme oxygenase 1; IPP, Isoprenoid pyrophosphate; LPCAT3, Lysophosphatidylcholine acyltransferase 3; Mevalonate, Mevalonic acid; MUFA, Monounsaturated fatty acid; NCOA4, Nuclear receptor coactivator 4; NF2, Neurofibromin 2; NOX4, NADPH oxidase 4; NRF2, Nuclear factor erythroid 2-related factor 2; P53, Tumor protein P53; PL-OOH, Phospholipid hydroperoxide; PUFA, Polyunsaturated fatty acid; PUFA-CoA, Polyunsaturated fatty acid coenzyme A; ROS, Reactive oxygen species; SCD1, Stearoyl-CoA desaturase-1; SREBP2, Sterol regulatory element-binding protein 2; System Xc-, Cystine/glutamate antiporter; TAZ, Transcription coactivator with PDZ-binding motif; TGase, Transglutaminase; TCA cycle, Tricarboxylic acid cycle; YAP, Yes1-associated transcriptional regulator.
System Xc⁻, a sodium-independent cystine/glutamate antiporter, consists of two subunits: SLC7A11 and solute carrier family 3 member 2 (SLC3A2) (85, 86). It exports glutamate while importing cystine, initiating intracellular GSH production (85). Inhibition of SLC7A11 triggers ferroptosis, and its expression is modulated by factors such as ubiquitin aldehyde-binding protein 1 (OTUB1), BECN1, p53, and activating transcription factor 3 (ATF3) (85).
The ferroptosis inducer erastin inhibits System Xc⁻, depleting GSH and promoting lipid peroxidation (11). Mutant p53 suppresses SLC7A11 expression, increasing lipid peroxide accumulation and inducing ferroptosis, although p53 can also upregulate GSH levels through p21 activation (87, 88). ATF3 and ATF4 regulate System Xc⁻ activity, either promoting or mitigating ferroptosis depending on context (89, 90). Nrf2 enhances SLC7A11 transcription, boosting GSH synthesis, while its loss amplifies ferroptosis susceptibility (91).
Phosphorylated BECN1 inhibits SLC7A11, inducing ferroptosis (92). In liver cancer, the ferroptosis-inducing drug sorafenib is effective at low doses but often encounters resistance (93, 94). DJ-1 suppression can enhance sorafenib efficacy (94). Additionally, dipeptidase-1 (DPEP1) exacerbates oxidative stress, increasing ferroptosis susceptibility (95).
GPX4, essential for lipid peroxide detoxification, relies on selenium and GSH. Selenium not only supports GPX4 expression but also plays a role in immune responses; its deficiency results in embryonic lethality (96, 97). The mevalonate pathway further regulates GPX4 expression and ferroptosis sensitivity (98, 99). Ferroptosis inducers such as Ferroptosis-Inducer-56 (FIN56) and RSL3 target GPX4 (87, 100). Additionally, ferroptosis suppressor protein-1 (FSP1), identified through CRISPR screening, inhibits ferroptosis independently of GPX4, highlighting the importance of the Coenzyme Q10 (CoQ10)–FSP1–NAD(P)H axis (101, 102).
In IRI, dysfunction of the System Xc⁻–GSH–GPX4 axis exacerbates oxidative stress and cellular damage. Reperfusion generates large amounts of ROS, while impaired System Xc⁻ function limits GSH replenishment, rendering GPX4 inactive. The resulting lipid peroxidation intensifies cell injury and ferroptosis. Similarly, in sepsis, extensive inflammation, oxidative stress, and immune activation disrupt this axis. System Xc⁻ inhibition reduces GSH availability, weakening cellular defenses against oxidative damage. Additionally, reduced GPX4 activity prevents lipid peroxidation resolution, leading to membrane instability and ferroptosis, which accelerates the progression of multiple organ failure.
4.5 CoQ10-FSP1-NADH axisCoQ10, also known as ubiquinone, is a lipophilic metabolite synthesized through the mevalonate pathway. Comprising an isoprenoid polymer and a benzoquinone ring, CoQ10 is indispensable for mitochondrial energy production (103). In its reduced form, it functions as a potent antioxidant, neutralizing free radicals generated by lipid peroxidation and thereby preventing ferroptosis (104).
CoQ10 is synthesized from acetyl-coenzyme A through the MVA pathway and is regulated at both transcriptional and translational levels (105). FSP1, initially identified as p53-responsive gene 3 (PRG3) and associated with p53-mediated apoptosis, shares sequence similarities with apoptosis-inducing factor (AIF) (106, 107).
FSP1, a flavoprotein oxidoreductase, interacts with DNA and uses NADPH to reduce ubiquinone to ubiquinol outside mitochondria, thereby inhibiting lipid peroxidation independently of GPX4 and GSH (101, 102). Elevated FSP1 expression enhances resistance to ferroptosis, a process regulated by murine double minute 2 (MDM2) and murine double minute X (MDMX) through peroxisome proliferator-activated receptor α (PPARα) (108). Plasma-activating mediators (PAM) reduce FSP1 expression, triggering ferroptosis in lung cancer, while the compound NPD4928 inhibits FSP1 and promotes ferroptosis (106, 107). Additionally, FSP1 reduces vitamin K to vitamin K hydroquinone (VKH2), which scavenges free radicals and prevents ferroptosis (109). The CoQ10-FSP1-NADH axis underscores FSP1 as a distinct ferroptosis inhibitor, separate from GPX4.
In IRI, the ischemic phase is marked by inadequate oxygen supply, reduced cellular metabolism, and diminished ATP and NADH production. Upon reperfusion, oxygen is reintroduced, rapidly generating ROS and inducing severe oxidative stress. FSP1 mitigates the expansion of lipid free radicals by reducing CoQ10 to panthenol using NADH, thus decreasing oxidative stress, limiting cell damage, and preventing ferroptosis. In sepsis, persistent inflammation and immune activation lead to excessive ROS production and enhanced lipid peroxidation. FSP1 function or expression may be impaired, exacerbating cellular antioxidant demands. The CoQ10-FSP1-NADH axis provides a crucial antioxidant pathway, removing lipid peroxides, maintaining membrane integrity, and reducing ferroptosis, thereby preventing multi-organ dysfunction to some degree.
4.6 DHODH-CoQH2 axisThe inner mitochondrial membrane hosts dihydroorotate dehydrogenase (DHODH), a key enzyme in pyrimidine ribonucleotide synthesis. Recent studies suggest that DHODH also functions as an antioxidant, similar to FSP1, by reducing CoQ10 and modulating ferroptosis independently of mitochondrial GPX4, highlighting its role in oxidative stress management and cell survival (110). DHODH catalyzes the oxidation of dihydroorotate (DHO) to orotate (OA) and facilitates the conversion of ubiquinone (CoQ) to ubiquinol (CoQH2), integral to respiratory complexes and electron transfer in oxidative phosphorylation (111). High DHODH expression confers ferroptosis resistance, while low expression increases susceptibility. DHODH functions independently of FSP1, working alongside GPX4 to prevent mitochondria-associated ferroptosis (110). Wang et al. demonstrated that DHODH provides an independent antioxidant pathway, complementing GPX4 in ferroptosis regulation (112). DHODH inhibitors, such as SA771726 (a leflunomide metabolite) and Brequinar sodium (BQR), are important in cancer therapy (113).
In IRI, the initial ischemic phase disrupts mitochondrial function and blocks the electron transport chain. The subsequent restoration of oxygen during reperfusion triggers the rapid generation of ROS. The DHODH-CoQH2 axis mitigates this damage by maintaining mitochondrial redox balance, reducing ROS-induced cellular damage, preventing lipid peroxidation, and inhibiting ferroptosis. In sepsis, sustained oxidative stress from systemic inflammation results in mitochondrial dysfunction and enhanced lipid peroxidation. The DHODH-CoQH2 axis alleviates oxidative stress, supports mitochondrial and cellular membrane integrity through continuous CoQH2 supply, and inhibits ferroptosis, thereby protecting cells and reducing organ dysfunction.
4.7 GCH1-BH4 axisTetrahydrobiopterin (BH4), synthesized by GTP cyclohydrolase 1 (GCH1) from guanosine triphosphate, plays a crucial role in limiting intracellular CoQ and ROS accumulation, preventing ferroptosis. It specifically inhibits the consumption of phospholipids containing polyunsaturated fatty acyl tails (114). While BH4 is essential for various enzymes, its role in ferroptosis inhibition is newly discovered. As an antioxidant, BH4 reduces lipid peroxidation, traps free radicals, and safeguards cells, particularly under conditions where GPX4 is inhibited. BH4’s protective effects are enhanced through regeneration by dihydrofolate reductase (DHFR) (115).
Kraft et al. demonstrated that GCH1 increases BH4 synthesis, facilitates lipid remodeling, and reduces lipid peroxidation, thereby inhibiting ferroptosis (114). Additionally, BH4 aids in converting phenylalanine to tyrosine, which is subsequently transformed into 4-OH-benzoate, a precursor for CoQ10 synthesis, further promoting ferroptosis inhibition. Studies underscore the GCH1-BH4 axis as critical in regulating tumor cell susceptibility to ferroptosis by modulating iron metabolism and mitigating ferritin toxicity through inhibition of NCOA4-mediated ferritin autophagy in colorectal cancer (115, 116). Moreover, Nrf2 has been shown to activate GCH1, promoting BH4 synthesis and protecting cells from oxidative stress, particularly in radiation-induced damage (117). The GCH1-BH4 axis thus represents a major regulatory pathway for ferroptosis, offering a strategy independent of the GSH/GPX4 system.
In IRI, the GCH1-BH4 axis enhances antioxidant defense by boosting BH4 synthesis, reducing ROS damage to cellular membranes, and preventing ferroptosis-induced cell death. In sepsis, this axis helps minimize cellular damage, stabilizes the intracellular environment, and reduces lipid peroxidation, thereby lowering the risk of organ dysfunction.
4.8 p53The tumor suppressor gene p53 plays a central role in managing cellular stress, including ribosomal stress, hypoxia, and DNA damage. Mutations in p53 and associated signaling pathways are key drivers of tumorigenesis (118). Ferroptosis, a process regulated by p53, is modulated through both transcription-dependent and independent mechanisms (119). p53 promotes ferroptosis by downregulating SLC7A11 expression through Ubiquitin-specific proteinase 7 (USP7), reducing histone H2B monoubiquitination (2). It binds to the SLC7A11 promoter, thereby affecting GPX4 activity and inducing lipid peroxidation (60). However, p53 does not significantly impact GSH levels or GPX4 function, likely due to the activation of Glutaminase 2 (GLS2), TIGAR (p53-induced glycolysis-regulated phosphatase), and CDKN1A (60). p53 activation increases expression of TfR1 and SLC25A28, leading to iron accumulation and heightened sensitivity to ferroptosis (69, 120).
Additionally, p53 targets Spermidine/spermine N1-acetyltransferase 1 (SAT1), enhancing ALOX15 activity and promoting lipid peroxidation under oxidative stress (121). In contrast, p53 can inhibit ferroptosis by enhancing GSH levels through the p53-p21 pathway and regulating ROS through Parkin (122). The p53-DPP4 interaction within the nucleus suppresses Dipeptidyl peptidase-4 (DPP4) and inhibits NADPH oxidase (NOX1), offering protection against ferroptosis (108). DPP4 inhibitors, such as vildagliptin, may reduce the anticancer efficacy of ferroptosis activators (88). MDM2 ubiquitinates p53, but MDM2 inhibitors like nutlin-3 can delay ferroptosis (122). p53 also inhibits erastin-induced ferroptosis by inducing CDKN1A in specific cell types (122). Overall, p53’s regulation of ferroptosis is complex, influencing lipid, energy, and iron metabolism in a cell-type and context-dependent manner.
In IRI, p53 enhances cellular susceptibility to ferroptosis by inhibiting antioxidant defenses and promoting lipid peroxidation. This process may be protective by clearing damaged cells in ischemia-reperfusion, but it can also exacerbate tissue damage and dysfunction. In sepsis, p53 similarly mediates oxidative stress-induced ferroptosis. Inflammation-driven persistent oxidative stress activates p53, which suppresses antioxidant systems and drives lipid peroxidation, thus triggering ferroptosis. While this cell death aids in clearing damaged cells and pathogens, excessive ferroptosis can damage tissues and organs, exacerbating sepsis-related organ failure.
4.9 Nrf2Cloned in 1994, Nrf2 is a member of the basic leucine zipper (bZIP) transcription factor family, including cap “n” collar (CNC) proteins. It is crucial for maintaining redox homeostasis and preventing oxidative stress by regulating genes associated with oxidative stress (123). During ferroptosis, Nrf2 activates cytoprotective genes involved in iron metabolism and redox signaling.
Nrf2 promotes the transcription of antioxidant-related genes by binding to antioxidant response elements (AREs) (124). Upon oxidative stress, Nrf2 dissociates from Kelch-like ECH-associated protein 1 (KEAP1) and translocates to the nucleus, triggering the expression of genes linked to downstream AREs. These include HO-1, quinone oxidoreductase 1 (NQO1), Fpn1, ferritin heavy chain 1 (FTH1), ferritin light chain (FTL), and Fpn1, which collectively promote GSH synthesis and limit ROS production (125, 126).
The Nrf2/KEAP1 pathway enhances the expression of System Xc⁻, which targets SLC7A11, indicating that SLC7A11 may mitigate Nrf2-mediated ferroptosis (127). Nrf2 has been shown to inhibit ferroptosis in mice with folate-induced kidney injury through the regulation of GPX4. It also facilitates NADPH production in the pentose phosphate pathway (99). Metallothionein 1G (MT1G), an Nrf2 target gene, helps hepatocellular carcinoma (HCC) cells resist ferroptosis (128). Additionally, Nrf2-driven ATP-binding cassette subfamily C member 1 (ABCC1/MRP1) promotes GSH efflux, potentially influencing ferroptosis regulation (129).
Nrf2 serves as a critical negative regulator of ferroptosis within complex molecular signaling networks. However, further preclinical and clinical research is essential to fully elucidate Nrf2’s role in ferroptosis resistance and the therapeutic potential of nuclear factor erythroid 2-like 2 (NFE2L2) inhibitors.
In IRI, Nrf2 mitigates ferroptosis-induced cell damage by upregulating antioxidant enzymes and related protective proteins, thereby reducing ROS accumulation and lipid peroxidation. This protective effect is crucial for minimizing tissue damage and facilitating repair after reperfusion. In sepsis, Nrf2 effectively reduces excessive ROS generation and lipid peroxidation, regulating antioxidant gene expression to preserve cell survival and function. Nrf2 activation stabilizes the intracellular environment, preventing multiple organ injuries induced by ferroptosis and improving sepsis prognosis.
4.10 YAP1/WWTR1The Hippo signaling pathway depends on two key effector proteins: WW domain-containing transcriptional regulator 1 (WWTR1/TAZ) and Yes1-associated transcriptional regulator (YAP1, also known as YAP), which control organ size, tissue homeostasis, and cancer growth (130, 131). Targeting YAP1/WWTR1 has therapeutic potential in cancer treatment and regenerative medicine (130, 131). These proteins act as biomechanical and morphological sensors, responsive to the cellular environment.
Cadherin 1 (CDH1/E-cadherin)-dependent cell adhesion enhances ferroptosis resistance through the Hippo pathway (55). E-cadherin activates the tumor suppressor neurofibromin 2 (NF2) in epithelial cells, reducing YAP1 activity and ferroptosis (55). Conversely, inhibiting Hippo pathway effectors such as NF2/merlin, large tumor suppressor kinases 1 (LATS1), and 2 (LATS2) reinstates cancer cell susceptibility to ferroptosis, particularly at high cell densities (55).
WWTR1 modulates angiopoietin-like 4 (ANGPTL4) and epithelial membrane protein 1 (EMP1) levels, while YAP1 influences transferrin receptor complexes (TFRC) and ACSL4 translation, promoting lipid peroxidation and iron accumulation, thereby inducing ferroptosis (55, 132). In conclusion, YAP1 and WWTR1 are key transcriptional regulators of ferroptosis, though further in vivo studies are necessary.
During IRI, YAP1/WWTR1 activation helps cells manage oxidative stress and attenuate ferroptosis by promoting antioxidant gene expression and defense mechanisms. Modulating YAP1/WWTR1 activity may alleviate tissue injury following ischemia-reperfusion and protect cells from lipid peroxidation and ferroptosis. In sepsis, YAP1/WWTR1 acts as a regulator, helping cells cope with excessive ROS generation and oxidative damage (133). Its activation promotes antioxidant gene expression, metabolic regulation, and reduces ferroptosis-associated apoptosis and histological damage, thereby preserving organ function and minimizing multiple organ damage caused by sepsis.
4.11 Activating transcription factorATF, part of the cAMP response element-binding protein 2 family, which includes ATF3 and ATF4, is critical for pathways involving autophagy, oxidative stress, and inflammation (134). Endoplasmic reticulum (ER) stress stimulates ATF expression, influencing metabolism and oxidative balance (135).
In human fibrosarcoma cells, ATF3 directly represses SLC7A11 during erastin-induced ferroptosis (89). Conversely, ATF4 activation enhances SLC7A11 expression, modulating ferroptosis (90). Overexpression of DNA damage-inducible transcript 3 (DDIT3/CHOP) in human glioblastoma, pancreatic, and Burkitt’s lymphoma cells links ATF4 to ferroptosis-associated diseases, such as Burkitt’s lymphoma and diabetic myocardial IRI (136, 137).
ATF4’s target gene, ChaC glutathione-specific γ-glutamyl cyclotransferase 1 (CHAC1), catalyzes GSH degradation, increasing the susceptibility of triple-negative breast cancer cells to ferroptosis (138). In contrast, heat shock 70 kDa protein 5 (HSPA5), an ER chaperone, mitigates ferroptosis by stabilizing GPX4 (139). These findings underscore ATF4’s involvement in ferroptotic cell death through complex signaling pathways, highlighting the need for further research on its precise role in ferroptosis (90).
In IRI, ATF4 is activated under ER stress and regulates cellular responses to protein misfolding, crucial for maintaining cellular function and preventing ferroptosis. Enhancing ATF4 activity has been shown to improve cellular tolerance to oxidative stress induced by ischemia-reperfusion (I/R), reducing cell death associated with ferroptosis (140). ATF4 also protects against oxidative damage and modulates immune responses in sepsis by regulating cellular redox balance. Its activation increases the expression of antioxidant proteins, alleviates lipid peroxidation, and prevents ferroptosis caused by excessive ROS accumulation, thereby safeguarding organs and tissues.
4.12 Hypoxia-inducible factorHIF plays a pivotal role in cellular responses to fluctuating oxygen levels. It is composed of a stable β component, aryl hydrocarbon receptor nuclear translocator (ARNT1/HIF1B), and a labile α subunit, endothelial PAS domain protein 1 (EPAS1/HIF2A), which together drive the expression of hypoxia-inducible factor 1α (HIF1A) and HIF3α, forming a heterodimeric transcription factor (141).
Under normoxic conditions, HIF1α and EPAS1 are hydroxylated by EGLN enzymes, which target them for degradation via the Von Hippel-Lindau (VHL) E3 ubiquitin ligase complex (142). HIF modulates ferroptosis in tumor cells through two mechanisms: EGLN serves as a key target for iron chelators like deferoxamine, which stabilize HIF by inhibiting EGLN (143).
Hypoxia induces HIF1α expression in HT-1080 fibrosarcoma cells, leading to increased transcription of fatty acid binding proteins 3 (FABP3) and 7 (FABP7), enhancing lipid uptake and storage, which inhibits ferroptosis (144). In contrast, EPAS1 upregulates hypoxia-induced lipid droplet-associated proteins (HILPDA/HIG2) in clear-cell carcinoma cells, promoting ferroptosis through lipid peroxidation and increased PUFAs synthesis (145).
These findings suggest that tumor type influences HIF’s regulation of ferroptosis. Understanding HIF’s role across different cancers could inform innovative therapeutic strategies for cancer treatment and prevention.
HIF is activated during ischemia in IRI, promoting cellular adaptation to hypoxic conditions and mitigating ROS damage by regulating antioxidant genes and metabolic pathways during reperfusion. This regulation inhibits ferroptosis. By controlling glycolysis and other metabolic pathways, HIF enhances cellular energy production and antioxidant capacity, which is critical for responding to hypoxia and oxidative stress. In sepsis, HIF modulates metabolic and inflammatory responses by regulating hepcidin and ferroportin expression, thereby maintaining intracellular iron homeostasis. This reduces free iron accumulation, limits excessive ROS generation through the Fenton reaction, and promotes antioxidant enzyme expression, aiding in ROS scavenging and preventing oxidative stress-induced ferroptosis.
5 Role of ferroptosis in critical illness5.1 IRIIRI is a common, life-threatening clinical complication with significant local and systemic effects. Recent studies suggest that ferroptosis plays a role in the pathophysiology of IRI across various organs, including the brain, intestines, liver, heart, and kidneys (20, 72, 146, 147). Modulating lipid peroxidation and iron levels influences susceptibility to ferroptosis, thereby affecting IRI outcomes (Figure 3).
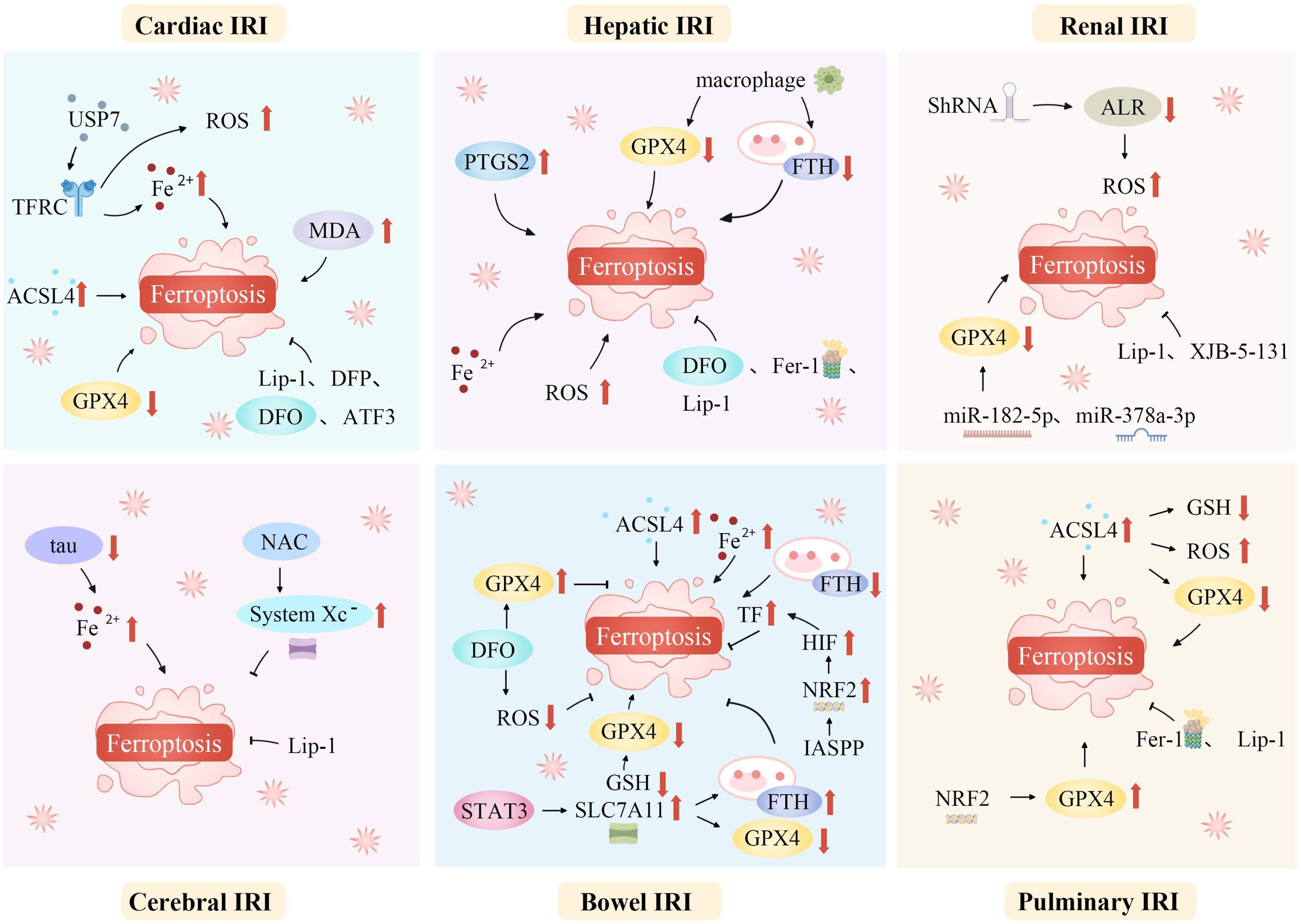
Figure 3. Ferroptosis mediates a variety of ischemia-reperfusion injuries. Ferroptosis, a unique regulated cell death form, mediates various IRIs. During ischemia, restricted blood supply causes hypoxia, disrupting metabolism and leading to intracellular changes like ROS generation in mitochondria’s electron transport chain. Reperfusion restores blood flow but worsens oxidative stress. Increased ROS overwhelms antioxidant systems like GPX4, triggering ferroptosis. Abundant tissue iron ions in the ROS-influenced Fenton reaction generate hydroxyl radicals, initiating lipid peroxidation. This is key in ferroptosis. In organs like the heart, brain, kidneys, and liver, ferroptosis-caused lipid metabolism disruption and peroxide accumulation damage cell membranes, causing cell death. Thus, ferroptosis-mediated damage in ischemia-reperfusion contributes to tissue dysfunction and organ failure. ACSL4, Acyl-CoA synthetase long-chain family member 4; ALR, Augmenter of liver regeneration; ATF3, Activating transcription factor 3; DFO, Deferoxamine; DFP, Deferiprone; Fe2+, Iron ion; Fer-1, Ferrostatin-1; FTH, Ferritin heavy chain; GPX4, Glutathione peroxidase 4; GSH, Glutathione; HIF, Hypoxia-inducible factor; IASPP, Inhibitor of apoptosis stimulating protein of p53; Lip-1, Liproxstatin-1; IRI, Ischemia-reperfusion injury; NAC, N-acetylcysteine; NRF2, Nuclear factor erythroid 2-related factor 2; PTGS2, Prostaglandin-endoperoxide synthase 2; ROS, Reactive oxygen species; ShRNA, short hairpin RNA lentivirals; SLC7A11, Solute carrier family 7 member 11; STAT3, Signal transducer and activator of transcription 3; System Xc-, Cystine/glutamate antiporter; TF, Transferrin; TFRC, Transferrin receptor complexes; USP7, Ubiquitin-specific proteinase 7.
5.1.1 Cardiac IRICardiac IRI and myocardial infarction are prevalent cardiovascular conditions associated with poor prognosis and limited early intervention options. Before the identification of ferroptosis, studies indicated that mitochondrial overexpression of GPX4 had cardioprotective effects against IRI (148). Ferroptosis predominantly occurs during the cardiac reperfusion phase, rather than during ischemia. Prolonged reperfusion results in a decline in GPX4 levels, while ferroptosis markers, including ACSL4, iron, and MDA, increase (149).
In 2015, Gao et al. first highlighted the role of ferroptosis in cardiac I/R (150). Iron concentrations increase near coronary blood flow after prolonged myocardial ischemia (151). Following cardiac IRI, cardiomyocytes become more vulnerable to ferroptosis as excess iron is absorbed by lipid peroxides through the Fenton reaction and enters the cells. Moreover, cardiomyocytes exposed to high iron levels become more susceptible to oxidative stress due to increased ROS, which, in turn, triggers ferroptosis.
Iron overload significantly contributes to myocardial cell injury, with lipid peroxidation playing a pivotal role in oxidative IRI (152, 153). In response to I/R, the heart exhibits increased production of ferritin heavy chain (FTH) and FTL, indicating elevated non-heme iron levels in ischemic myocardial tissue (72). Cardiomyocytes treated with erastin or RSL3 show decreased Fe2+, ROS, MDA, and cell death levels when ATF3 is overexpressed, preventing ferroptosis in these cells (152).
An in vitro study revealed that phospholipid oxidation products negatively affect cardiomyocyte viability during I/R (154). Oxidized lipidomic analysis identified oxidized phosphatidylethanolamine as a specific indicator of ferroptosis, providing direct evidence for cardiac ferroptosis (147). Moreover, I/R-treated hearts demonstrated the formation of oxidized phosphatidylethanolamine in their mitochondria, underscoring the role of cardiac mitochondria in lipid peroxide generation and ferroptosis signaling (155). Additionally, inhibiting or knocking down USP7 suppresses TFRC expression, reducing myocardial IRI primarily by decreasing iron levels, lipid peroxidation, and ferroptosis (120).
Li et al. investigated the in vivo inflammatory responses triggered by IRI following heart transplantation. Their results demonstrated that ferroptosis facilitated endothelial cell adhesion in cardiac tissues via the type I interferon/TLR4/Trif signaling pathway, which in turn promoted neutrophil recruitment to the injured myocardium (147). Moreover, using a coronary artery ligation model to induce myocardial IRI, they found that suppression of ferroptosis improved heart function and reduced infarct size.
5.1.2 Hepatic IRIHepatic IRI poses a significant challenge in liver transplantation, as it induces an immune response and inflammatory cascade that impairs donor liver function and worsens recipient prognosis (154). In pediatric living donor liver transplants (LTx), donor iron burden has been identified as an independent risk factor for liver IRI (156). Iron overload, lipid peroxidation, and the upregulation of Prostaglandin-endoperoxide synthase 2 (PTGS2)—a marker of ferroptosis—are key features of IRI progression in the liver.
In vitro studies have shown that hepatic macrophages play a role in ferroptosis during IRI, with macrophage co-culture leading to ferroptosis through downregulation of GPX4 and FTH1 (157). Additionally, mice subjected to a high-iron diet exhibited aggravated IRI, which was mitigated by treatment with DFO104.
5.1.3 Renal IRIFerroptosis is firmly linked to renal IRI, where it significantly contributes to the pathophysiological processes associated with the condition. The tubular manifestation of ferroptosis plays a crucial role in the progression of renal IRI (158).
In the context of renal injury and repair, augmenter of liver regeneration (ALR) has been shown to influence ferroptosis (159). Blocking ALR with short hairpin RNA (shRNA) exacerbates the condition, increasing ROS production, mitochondrial damage, and ferroptosis (159). Using a rat model of renal IRI, Ding et al. demonstrated that miR-182-5p and miR-378a-3p directly bind to the 3′-untranslated region of GPX4 mRNA, suppressing GPX4 expression and promoting ferroptosis (160).
Reducing iron-related antioxidants and administering iron chelators can improve renal function in patients with renal IRI (161). These findings underscore the close association between ferroptosis and renal IRI, involving multiple regulatory mechanisms.
5.1.4 Cerebral IRIThe pathophysiological mechanisms underlying cerebral IRI are multifactorial, involving factors such as heightened inflammatory responses, calcium overload, oxidative damage, excessive release of excitatory amino acids, and processes leading to apoptosis or necrosis (162). Most research on ferroptosis in cerebral IRI focuses on oxygen-glucose deprivation/reoxygenation models and animal models of focal or global brain injury.
In focal cerebral IRI, a middle cerebral artery occlusion (MCAO) model, based on the traditional MCAO technique, revealed reduced ferritin expression. In this model, ferritin overexpression modulated p53/SLC7A11-mediated ferroptosis, improving memory and cognitive function (163). Significantly suppressed tau expression, characteristic of cerebral I/R, is linked to Alzheimer’s disease and promotes iron efflux. This impairs iron transport to the extracellular space, increases intracellular iron levels, and induces neurotoxicity and ferroptosis, thereby exacerbating neuronal injury (164, 165).
Due to the complex nature of cerebral IRI and the incomplete understanding of ferroptosis mechanisms, it is critical to consider the interplay between multiple factors when studying the effects of ferroptosis on cerebral IRI. Results from single-perspective studies often fail to yield conclusive outcomes in broader animal models or clinical settings.
5.1.5 Bowel IRIThe intestine, particularly the small intestine, is one of the most vulnerable organs to IRI, which typically results from a sudden decrease in blood flow followed by reoxygenation upon restoration of circulation (165). This condition is associated with various forms of regulated cell death (RCD), including ferroptosis, apoptosis, necroptosis, and autophagy (166, 167). Lipid peroxidation and ROS generation play a pivotal role in initiating and completing ferroptosis in intestinal IRI (168).
A crucial enzyme, ACSL4, regulates lipid content and is vital for initiating ferroptosis (57). Li et al. demonstrated that intestinal IRI leads to decreased levels of GPX4, ferritin heavy chain, and GSH, while increasing ACSL4 and iron levels. This process results in ferroptosis of intestinal histiocytes (146). Inhibiting ACSL4, a key ferroptosis controller, through medicinal and biological approaches successfully prevented lipid peroxidation and ferroptosis. This intervention also alleviated cell injury and dysfunction of the intestinal barrier caused by IRI (57, 146).
Ferroptosis in intestinal IRI has been further investigated through modulation of SLC7A11, which influences intestinal IRI-induced acute lung injury (ALI) (169). The inhibitor of apoptosis-stimulating protein of p53 (IASPP) mitigates ferroptosis in intestinal IRI by activating the Nrf2-HIF1-TF signaling pathway, thereby reducing ALI (170).
Additionally, intestinal microbiota and its metabolites can inhibit ferroptosis in intestinal IRI. A potential mechanism involves capsaicin ester, a metabolite of the microbiota, which activates transient receptor potential cation channel subfamily V m
留言 (0)