Design and Fabrication of Scaffolds
Scaffolds were designed using the computer-aided design (CAD) software SolidWorks (Dassault Systèmes) with emphasis on porosity and pore size. The scaffolds were designed to mimic bone’s natural trabecular and cortical architectures. The central trabecular section of the scaffold was designed with alternating horizontal and vertical layers, with a layer height of 0.5 mm and each layer having channels formed by 0.5-mm-wide struts spaced 0.5 mm apart. This design allows for horizontal and vertical flow paths and a highly porous cross-section mesh with square holes (0.5 × 0.5 mm). The cortical section was designed with long, hollow cylindrical structures representing bone osteons. Individual cylindrical structures were arranged in concentric layers around the central trabecular section. These structures were 3 mm in diameter with a wall thickness of 0.5 mm. Cortical wall porosity of approximately 50% was achieved by creating 300-µm pores in the walls, along both the length and the circumference of the structures. Cura software (Ultimaker) was used to define print parameters such as layer height and infill percentage, followed by slicing the designs to generate the G-code. The Ultimaker 2 + 3D printer was used to print the scaffolds with PLA filament purchased from Ultimaker and Gizmo Dorks (2.85-mm filament diameter, MW 60,000–80,000). The scaffolds were printed using a 0.25-mm-diameter print nozzle to achieve the best possible resolution.
Mechanical Characterization
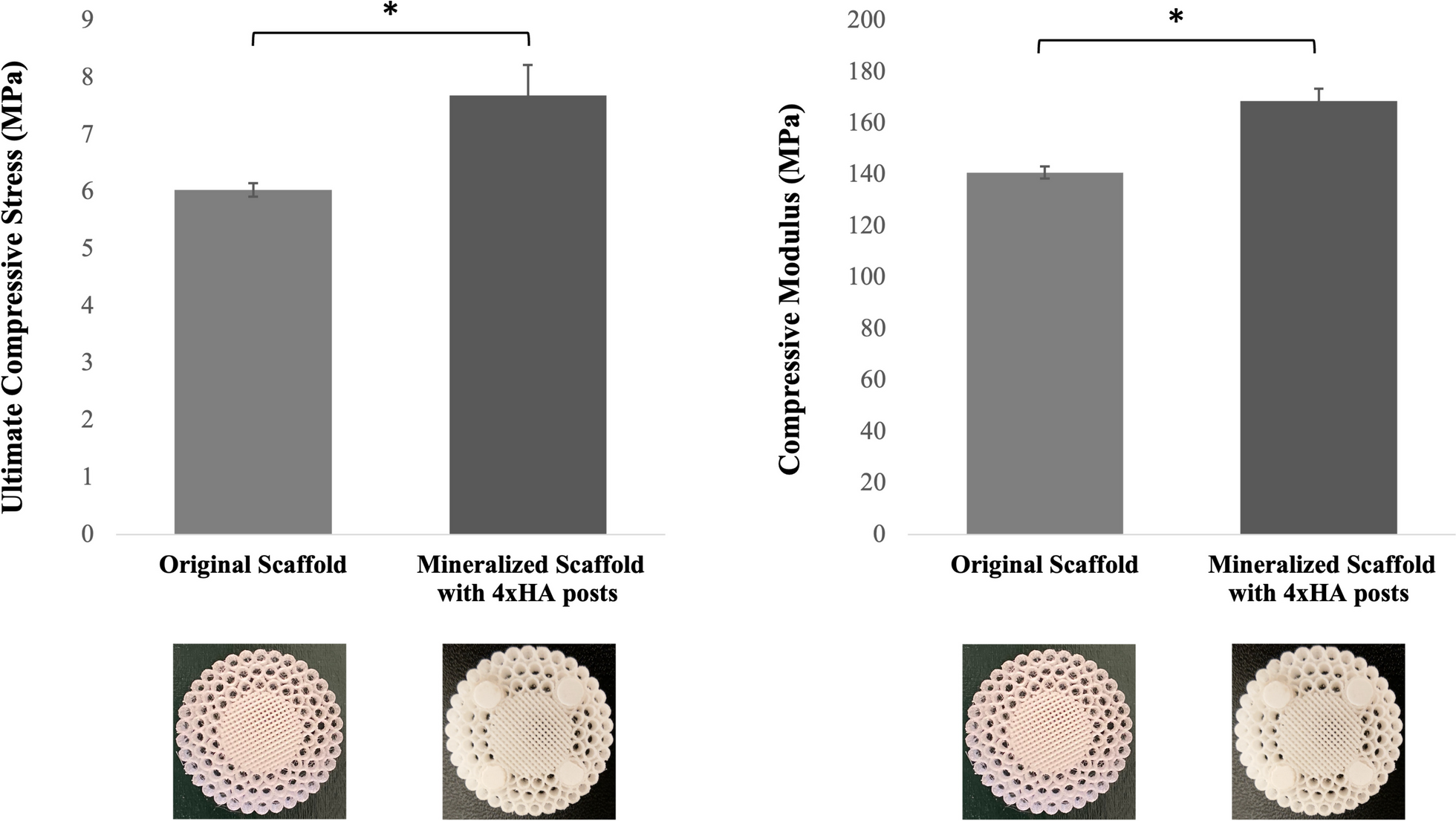
Compression analysis was performed to determine the scaffold’s mechanical strength. Whole scaffolds (36 mm in diameter, 10 mm in height) with an inner trabecular Sect. (18 mm in diameter) and an outer cortical Sect. (3 concentric layers) were used. Scaffolds (n = 3) were compressed at a uniform strain rate of 1 mm/min (10% strain/min) using the Instron 5869 mechanical testing system. The data generated was analyzed to determine the compressive modulus and ultimate compressive strength. Four HA posts (6 mm in diameter and 10 mm in height) were incorporated into the scaffold; previous studies conducted in our lab on nanofibrous scaffolds showed that the incorporation of HA posts within the cortical section significantly improved the scaffold’s mechanical properties [22]. For this purpose, scaffolds were designed and 3D printed with hollow cylindrical portions within the cortical sections to incorporate these posts. The HA posts were developed in Dr. Cook Chennault’s lab at Rutgers University. Briefly, the posts were made of 100% HA (density 3.14 g/cm3) with a PVA binder (1.269 g/cm3). The HA was first ball milled for 6 h and then added to PVA dissolved in water (5:95). This was magnetically stirred for 2 h and then dried for 2 h before placing it in a micromill for 10 s. The HA was then placed into a die and cold-pressed at ambient temperature for 15 min. Finally, the posts were sintered at 1300 °C for 2 h. These individual HA posts were also tested under the same conditions of compression to determine their mechanical characteristics. The number and arrangement of these posts were based on results from a finite element analysis (FEA) that was previously conducted in our lab [22]. Scaffolds with HA posts were subject to compression testing and their mechanical properties were compared to those of the scaffolds with no posts.
Mineralization in Concentrated Simulated Body Fluid (SBF)
Scaffolds were mineralized in concentrated SBF (10 × SBF) as previously described by Andric et al. (2015) [20], Andric et al. (2011) [21], and Tas et al. (2004) [23]. In this process, calcium ions from simulated body fluid (SBF) attach to nucleation sites on the polymer surface. SBF has a calcium concentration similar to blood plasma, and the addition of the inorganic content in the form of Ca2+ ions improves the scaffold’s osteoblastic differentiation capability [24]. Static mineralization of scaffolds was carried out in 10 × SBF. The mineralization procedure involved placing scaffolds in vials filled with 100:1 SBF and NaHCO3 and placing the vials on a shaker [23]. The solutes used in the simulated body fluid (SBF), sodium chloride (NaCl), potassium chloride (KCl), calcium chloride dihydrate (CaCl·H20), magnesium chloride heptahydrate (MgCl2·H20), sodium bicarbonate (NaHCO3), and sodium phosphate monobasic (NaH2PO4) were purchased from Fisher Scientific (Pittsburgh, PA, USA). Mineralization was carried out for 10 h with the 10 × SBF solution changed every hour.
Mineral Degradation
The degradation of the mineral coating on the scaffolds was studied over 9 weeks. For the purpose of this study, solid cylindrical scaffolds (radius 2 mm, height 8 mm, and weight 0.114 ± 0.001 g) were 3D printed and mineralized as discussed in the previous section. To study mineral degradation, the scaffolds (n = 4) were placed in 15-ml tubes filled with 10 ml of phosphate-buffered saline (PBS) (pH 7.4). The tubes were placed on a shaker in an incubator set to 37 °C. The PBS solution was replaced once a week. At week 0, week 1, week 2, week 5, and week 9, the scaffolds were removed and rinsed with DI water before allowing to dry. The amount of mineral remaining on the scaffolds at the different time points was estimated and compared using Alizarin red staining (Alizarin red powder, Sigma-Aldrich). Briefly, scaffolds were incubated with a 40-mM Alizarin red solution for 10 min. They were washed five times with DI water, washed once with PBS, and incubated with PBS for 15 min. PBS was removed, and cetylpyridinium chloride (CPC) (Sigma-Aldrich) was added to the scaffolds. Stain controls were made using CPC and Alizarin red stain, with a decreasing concentration curve 4 mM, 1 mM, 0.25 mM, 0.025 mM, 0.0025 mM, 0.00025 mM, and 0.000025 mM. Triplicates of 100 μl samples were removed from standards as well as scaffolds and put in a 96-well plate. Absorbance was measured at a wavelength of 652 nm.
Creating Additional Micropores
Scaffolds must be highly porous in order to support bone formation and the formation of its vasculature. In addition to the pores present in the scaffold design, additional pores were introduced using a soak-freeze technique. For this study, trabecular sections (long radius 4.5 mm, short radius 3.9 mm, height 5 mm) were first soaked in DI water overnight. After removing excess water, the scaffolds were frozen by placing them in liquid nitrogen or in the freezer at − 80 °C overnight. To check for the presence and structure of soak-freeze induced pores, scanning electron microscopy (SEM) was used. Images of the scaffold surface were obtained post the different freezing methods and compared to images obtained from scaffolds that were not subjected to any soak-freezing. In addition to this, liquid extrusion porosimetry (LEP-1100A, PMI, Ithaca, NY, USA) was used to determine and compare the pore size distributions produced as a result of the different freezing methods.
Evaluating Scaffold Biocompatibility
Scaffolds, 5 mm in diameter and 1 mm in height (trabecular design), were 3D printed and their biocompatibility was evaluated by studying cell adhesion and proliferation over the course of 2 weeks using bone marrow-derived human MSCs (HMSCs). Prior to seeding cells on the scaffolds, they were sterilized by soaking in 70% ethanol for 30 min, followed by UV radiation applied to both sides of the scaffolds for 15 min each. After sterilization, scaffolds were washed with PBS and preconditioned in cell culture media overnight. HMSCs were seeded at a cell density of 15,000 cells per scaffold or tissue culture plastic (TCP). They were grown and maintained in Alpha-Modified Minimum Essential Media (α-MEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Metabolic activity of cells was measured using PrestoBlue® assay, an indirect measurement of cellular viability at days 1, 3, 7, 10, and 14. Absorbance was measured at 570 nm and normalized using the initial cell number.
Scaffold Preparation for In Vitro Analysis
Scaffolds, with both the trabecular and cortical portions, were prepared for the in vitro studies as follows. The cortical sections which will be pre-vascularized later were filled with alginate (8% w/v) and cross-linked with 0.1 M calcium chloride solution. This was done to block the cortical sections before mineralization in 10 × SBF and to prevent any mineral deposition that could hinder vascular endothelial cell attachment [25]. The scaffolds were then mineralized in 10 × SBF as described previously. After mineralization, the alginate was removed, and the scaffolds were sterilized in 70% ethanol for 30 min followed by exposure to UV light for 30 min. The cortical portions of the scaffolds were then pre-vascularized as described below.
Pre-vascularization of the Cortical Section
In order to promote angiogenic and vasculogenic differentiation of MSCs, the cortical sections of the scaffolds were pre-vascularized by seeding with HMEC-1 s (ATCC). HMEC-1 s (passages 4–5) were seeded within the cortical sections. The medium used for this study was MCDB 131 medium (Life Technologies) with 1 µg/ml hydrocortisone, 10 mM glutamine, 10% FBS, 1% penicillin–streptomycin, and epidermal growth factor or EGF (10 ng/ml). In a preliminary study, the cells were seeded at a high seeding density of > 100,000 cells/cm2 and allowed to grow for 21 days. PrestoBlue® assay was performed on days 3, 7, 12, 14, and 21 to understand how the cells perform. In the final study, they were seeded at a cell density of 50,000 cells/cm2 and allowed to grow for 14 days, with the media changed every other day. PrestoBlue® assay was again used to check for cell viability on the scaffolds and the control group (TCP) at days 4, 8, 11, and 14. Scaffolds (n = 4) and cells on TCP were also fixed using 4% paraformaldehyde (PFA, Sigma-Aldrich) for CD31 staining. The remaining scaffolds were decellularized using a freeze–thaw method to leave behind a pro-angiogenic matrix. Briefly, the scaffolds were first placed in liquid nitrogen for 10 min, then in a water bath maintained at 37 °C for 10 min. The scaffolds were washed with PBS (3 ×) before repeating the freeze–thaw cycle two more times. Post-decellularization, the scaffolds were stored at − 80 °C.
Studying Scaffold Osteogenic and Angiogenic Differentiation Capabilities
Scaffolds were seeded with HMSCs. Two groups of scaffolds, the mineralized pre-vascularized (PV) scaffolds and mineralized non-vascularized (NV) scaffolds, were studied and compared to cells seeded on tissue culture plastic (TCP). The cells were seeded at P3 and at a seeding density of 15,000 cells/cm2. Cells used for this study were first grown in osteogenic media—the Mesenchymal Stem Cell Growth Kit, with Mesenchymal Stem Cell Basal Medium, l-alanyl-l-glutamine, 30% FBS, rh-IGF, rh-FGF, 1% penicillin–streptomycin (ATCC). When seeding on scaffolds and subsequent feeding, the media used were standard osteoblast media without any osteogenic factors—α-MEM (Life Technologies) with 10% FBS and 1% penicillin–streptomycin. Metabolic activity was monitored using the PrestoBlue® assay. Media were collected from all groups (n = 8) and cells were fixed on days 4, 8, and 12. Enzyme-linked immunosorbent assays (ELISA) were performed to study the differentiation of the HMSCs. Media collected were tested for the bone marker OC and the endothelial marker VEGF-A. The ELISA kits were purchased from RayBio®. Samples fixed at days 4, 8, and 12 were also stained for alkaline phosphatase (ALP), an early marker for osteogenesis using an ALP staining kit (Sigma-Aldrich). Immunohistochemical staining for CD31 was also performed. The CD31 primary monoclonal antibody and the secondary antibody were purchased from Thermo Fisher Scientific. All the images were obtained using a Zeiss confocal microscope.
Statistical Analysis of Data
All data have been presented as mean ± standard deviation. One-way analysis of variance (ANOVA) was used to determine if the differences between the groups were significant. A p-value < 0.05 was considered to be significant.







留言 (0)