2.1 KARS expression patterns in lung adenocarcinoma
To provide better functionalities they implemented a TIMER database comparing KARS gene expression patterns across various malignancies against normal tissue data. Our aim was to identify original KARS expression profiles in the context of different oncological diseases. Therefore, we further detected KARS expression in LUAD tissues and adjacent normal tissues using the TCGA 'LUAD' database [12, 13]. Based on six molecular immune subtypes determined by previous research, tumor samples were divided into high and low expression groups using the median values. The proportions of each subtype in each group were calculated, and the significance was tested using the chi-square test. Spearman correlation analysis was conducted to calculate the correlation between the KARS gene and the Tumor Immune Phenotype (TIP) score, as well as the autocorrelation of TIP scores, visualized using the linkET package. Wilcoxon rank-sum test was used to perform significance tests for differences between the normal group and tumor group for each tumor. RNA-seq data were obtained from the PanCanAtlas-provided normalized TCGA dataset, processed and normalized through the Firehose pipeline, and transformed into Z-Score values to enhance data comparability.
2.2 GSVA analysis
Samples were divided into high and low expression groups based on gene expression levels. Differential analysis was performed using the limma package to calculate log2 fold change (log2FC). Gene set enrichment analysis was carried out using the fgsea package to determine significance and visualize the results. Single-cell analysis data were processed through the CancerSEA website to define 14 functional states. The z-score algorithm in the GSVA package was used to calculate the activity of gene sets corresponding to functional states, and Pearson correlation analysis was employed to assess the correlation between gene expression and functional states. The GSVA package was utilized to score 73 KEGG metabolic gene sets and compare the metabolic GSVA scores between the high and low expression groups [14, 15].
2.3 Wilcoxon rank sum tests
Our data sources include the normalized TCGA dataset and the GSE40791 dataset. The RNA-seq data of TCGA is sourced from the file EBPlusPlusAdjustPANCAN_IlluminaHiSeq_RNASeqV2.geneExp.tsv provided by the PanCanAtlas project, and these data are generated using the MapSplice and RSEM algorithms through the Firehose analysis pipeline, followed by normalization. The probe matrix of the GSE40791 dataset is annotated as a gene matrix, and if a gene corresponds to multiple probes, the average of the expression levels of these probes is taken. The data from both datasets are transformed into Z-Score values to eliminate the influence of different dimensions, and Wilcoxon Rank Sum Tests are used to compare the expression level differences between tumor and normal tissues [16].
2.4 Genomic copy number variation
We used a CNV analysis method based on the gistic score to study the copy number profiles of tumors in the TCGA-LUAD project. Through processing and analyzing a large-scale sample data, we obtained CNV data from 516 samples. The data was visualized using bar graphs, with color coding to differentiate different ranges of gistic score scores. To investigate the correlation between copy number variation scores and gene expression levels, we utilized scatter plot analysis combined with the Spearman rank correlation coefficient method, which is a non-parametric statistical method used to measure the monotonic relationship between two variables. The copy number profiles were measured through whole-genome microarray and gene-level copy number estimates were generated using the GISTIC2 method. Subsequently, differences in gene expression among 5 types of copy number were compared, with significance testing conducted using the Kruskal–Wallis test [17, 18].
2.5 Expression of immune regulatory molecules in different states of KARS
We accessed the TISIDB website, which includes immunomodulators and chemokines, such as immune stimulating genes, immune inhibitory genes, chemokines, and human leukocyte antigens. We used the Wilcoxon rank-sum test to analyze the expression differences of immune-related molecules between the high and low expression groups of PDCD1, and visualized the average expression level of each gene in the two groups through a heatmap. Immune regulatory molecules are crucial for cancer immunotherapy. We studied the expression of immune regulatory molecules, somatic copy number alterations (SCNA), and expression control patterns through epigenetics to further understand the expression and control patterns of immune regulatory molecules in different states of KARS [19].
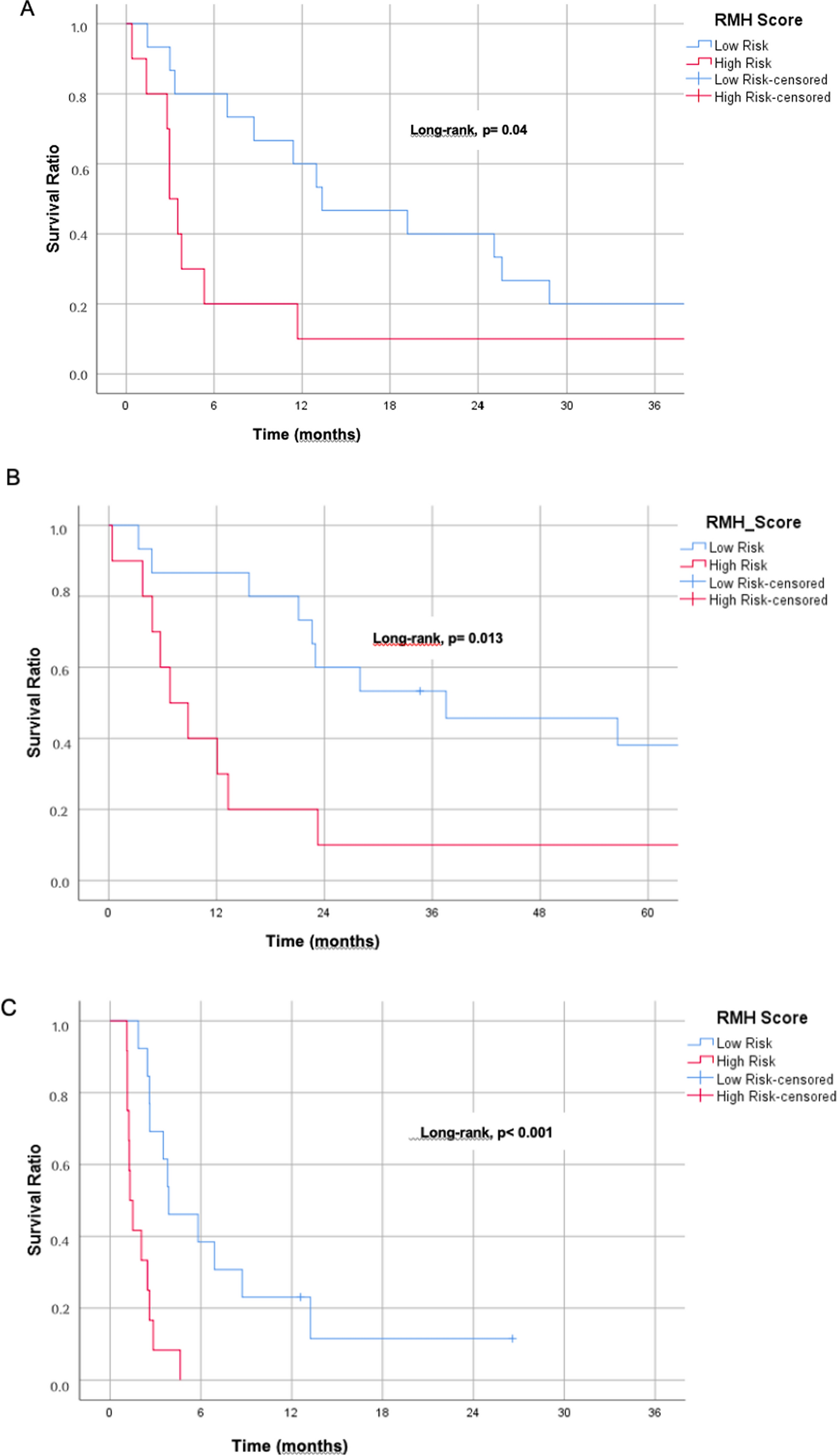
2.6 Survival analysis
In R, we performed Kaplan–Meier survival analysis using the survival package. We used the survminer package to identify high and low expression groups by finding the best cutoff values with minimum proportion of each group greater than 0.3. Survival differences were assessed by a Log-rank test with the survfit function for both groups. The inverse variance method was used to combine the results of single-factor Cox survival analysis for meta-analysis on hazard ratios. HR values of less than 1 were grouped together as representing anti-cancer effects, while those with HR greater than 1 were grouped as pro-cancer. We employed the meta package in R [17, 18], to run the statistical analysis and visualization [20, 21].
2.7 Meta-analysis of Cox analysis
First of all, meta-analysis was carried out on the logarithm value of HR as the main measurement index accompanied by single-factor Cox survival analysis through adopting an inverse variance method. Finally, we set the HR values as less than 1 (the anti-cancer effects) and greater than one representing pro-tumorigenesis. Statistical analysis and visualization were implemented in R (version 4.3.2) with the Meta package. We used the “survival” package to perform single-factor and multi-factor Cox survival analysis on genes and traditional clinical variables, described the relative risks, and used the “forestploter” package for visualization.
2.8 Single-cell analysis
The single-cell RNA sequencing (scRNA-seq) data of lung adenocarcinoma (LUAD) was extracted from the TISCH database specifically from the GSE131907 dataset. This data was analyzed to identify and characterize distinct cell populations expressing the KARS gene. Gene expression data is normalized to account for differences in sequencing depth and other technical variations. Techniques such as PCA (Principal Component Analysis) and t-SNE (t-distributed Stochastic Neighbor Embedding) are used to reduce the dimensionality of the data, facilitating visualization and interpretation. Cells are grouped into clusters based on their gene expression profiles using algorithms like Louvain clustering. This helps identify distinct cell populations within the tumor. Identify cells with significant expression of the KRAS gene, and analyze their distribution across different clusters. Pathway analysis and gene set enrichment analysis are conducted to explore the biological functions and pathways active in KRAS-expressing cells [22].
2.9 Functional enrichment analysis
Utilized the limma toolkit sourced from Bioconductor to carry out a differential gene expression analysis, allowing us to ascertain the log2 fold change (log2FC) for every gene. Post-analysis, we organized the genes based on their log2FC figures to identify those displaying the most significant expression variations. Subsequently, we employed the fgsea package [23].
2.10 RNA extraction
The manufacturer's instructions were followed to extract total RNA from the A549 cells and 16HBE with TRIzol reagent (Invitrogen, Carlsbad, USA). After RNA extraction, the concentration of total planted RNAs was assessed in nanogram on a Biophotometer spectrophotometer (Eppendorf; Germany). After that, cDNA synthesis was performed according to the manufacturer's protocol using TAKARA cDNA Synthesis Kit (TAKARA, Japan).
2.11 Reverse transcription-polymerase chain reaction (RT-PCR)
The cells were cultured, and total RNA was extracted using the manufacturer protocol for the RNAfast200 kit. Following this, 1 μg of RNA was used for reverse transcription (RT) to produce cDNA from which the template used in RT-PCR. Following IFN-γ stimulation, RT‑PCR was performed using the RevertAid First Strand cDNA Synthesis Kit. For all reactions PCR conditions were: an initial denaturation at 94 °C for three minutes, followed by thirty cycles of a denaturation step at 94 °C for thirty seconds, annealing was performed at 52°for different waiting times), extension (72 °C/ one min) and final extension cycle (five more minutes). The PCR conditions used were as follows: an initial denaturation at 94 °C for three minutes, followed by 30 cycles of denaturation at 94 °C for 30 s, annealing at 58 °C (beta-actin), or otherwise the calculated optimal temperature range) degrees Celsius dependent on gene-specific primer design using a step-gradient ranging from one degree below to two degrees above those temperatures then run again with Touchdown in tandem with another cycle-range gradient targeting maximum product yield and specificity taking into account each DNA-pair's melting point profile. Traditional gel electrophoresis visualized the gel images of PCR products. The FH mRNA expression levels in the A549 and 16HBE cell lines are analyzed by Gel-Pro software, with β-actin as an internal control. Experiments were done in triplicate [24]. KARS:F-GATCACCTGACTGACATCACCT;R-TACCAGGATTCCCCTGAACTC.MRPL12:F-ATCCCCATAGCGAAAGAACGG;R-GGACGAGGTTGATGCCTTGG.RPS27:F-ATGCCTCTCGCAAAGGATCTC;R-TGAAGTAGGAATTGGGGCTCT. RPLP13: F-GAGTCTGACGTTGGTGAGC; R-CAGTTCACCTCAGCAGCAG.
2.12 Statistical analysis
Continuous data were evaluated using either the Students t-test or the Wilcoxon rank-sum test, chosen based on the distribution characteristics of the data. Spearman’s rank correlation analysis was employed to assess relationships between variables. Statistical significance was determined for results with a p-value below the 0.05 threshold.





留言 (0)