2.1 Data collection and preprocessing
To explore the potential association between KLHDC8A expression and clinical prognosis of glioma patients, we retrieved data from several databases: CGGA, TCGA and GTEx. The raw expression data were downloaded, and normalization was performed using R (version 4.3.0). Specifically, expression levels from the GTEx and TCGA databases were log2-transformed and adjusted for batch effects using the sva package to ensure comparability across different platforms. Missing values in the clinical and expression data were imputed using the K-nearest neighbors algorithm implemented in the impute package in R. We analyzed KLHDC8A expression across various glioma subtypes and conducted differential expression analysis between normal and glioma tissues using the limma package. The Benjamini–Hochberg method was employed to determine statistical significance by adjusting p-values. Expression matrices for both healthy and cancerous tissues can be found in these databases.
2.1.1 Pan-cancer analysis of KLHDC8A expression
The pan-cancer expression profile data were downloaded from the UCSC Xena database (https://xenabrowser.net/datapages/), which includes normalized RNA-Seq data from TCGA across multiple cancer types. Expression data for KLHDC8A was extracted for all available tumor types, along with corresponding clinical metadata such as overall survival (OS) and disease-specific survival (DSS). To assess KLHDC8A expression across different cancer types compared to normal tissues, we conducted Wilcoxon rank sum tests and signed rank tests. This non-parametric approach was chosen to accommodate the potentially non-normal distribution of gene expression data. For several cancer types, we evaluated the expression levels in tumor and normal tissues, applying the Benjamini–Hochberg false discovery rate (FDR) correction to adjust for significance discrepancies. We constructed Cox proportional hazards regression models for overall survival (OS) and disease-specific survival (DSS) to examine the prognostic value of KLHDC8A. These models were adjusted for relevant clinical covariates, including age, gender, tumor stage, and treatment status. Separate models were fitted for each cancer type to explore cancer-specific associations.
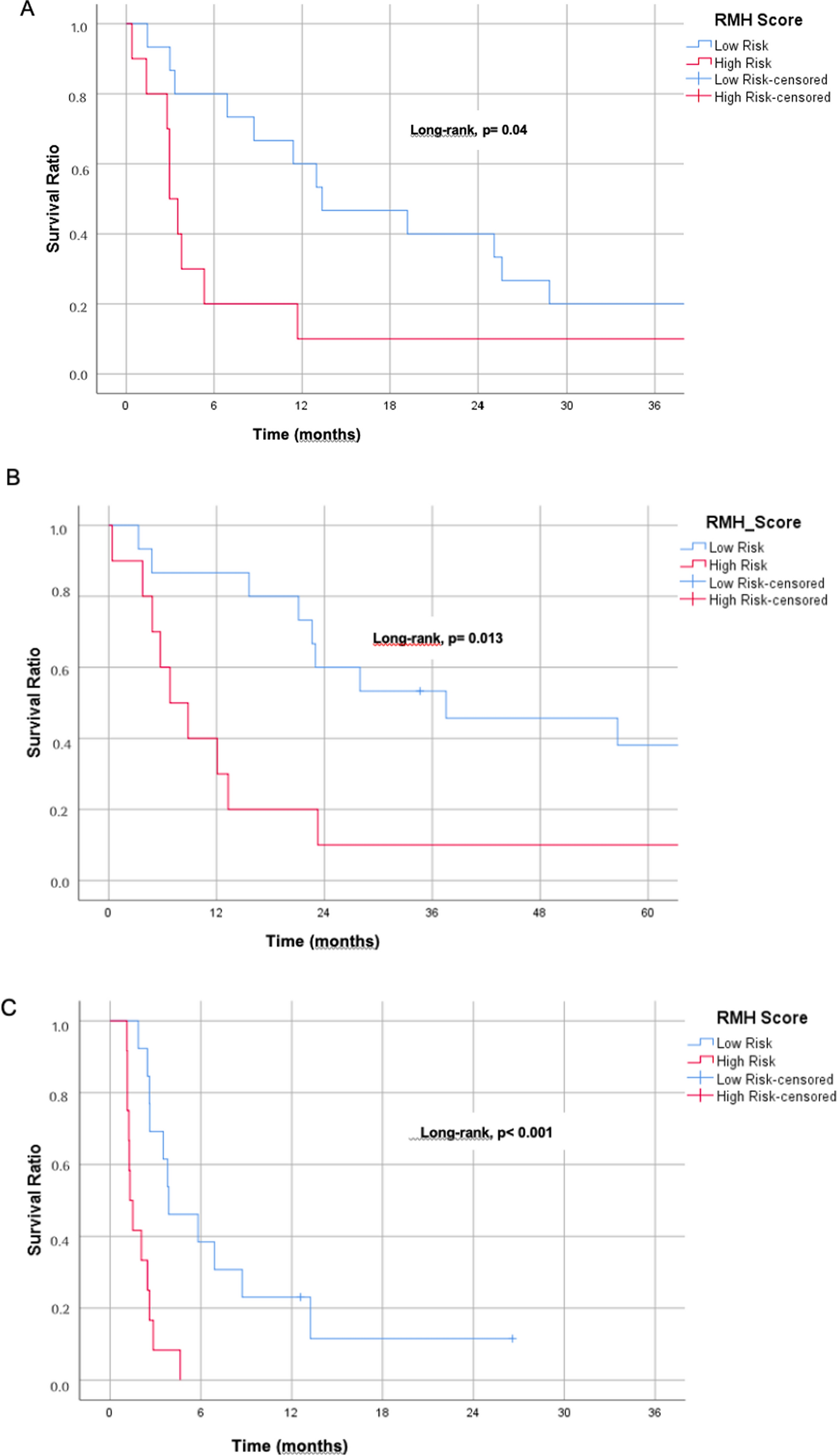
2.1.2 Prognostic analysis
Kaplan–Meier survival curves were created to evaluate the predictive importance of KLHDC8A expression in glioma patients. According to the median KLHDC8A expression level in the glioma group, patients were categorized as either "high" or "low" expression. In order to compare the two groups' survival results, including OS, the log-rank test was employed. At p < 0.05, a significant level was established. To quantify the relative risk of death associated with greater KLHDC8A expression, hazard ratios (HR) with 95% confidence intervals (95% CI) were included with the Kaplan–Meier analysis. Using Cox proportional hazards models, the HR was estimated. Additional evaluation of KLHDC8A expression's predictive potential in glioma prognosis was conducted using ROC curve analysis. Evaluation of KLHDC8A expression as a predictor of overall survival after five years was done using the ROC curve. To evaluate the discriminatory power of the model, we calculated the area under the ROC curve (AUC), where values closer to 1.0 indicate better sensitivity and specificity. The independence of KLHDC8A as a predictive factor was confirmed through univariate and multivariate Cox regression analyses.
2.1.3 Cell culture and cell transfection
We got the normal astrocyte cell line, human glioma cell lines TG905, LN308, T98, and U251 from the American Type Culture Collection (ATCC, Manassas, VA, USA). Once received, every cell line was grown in DMEM (Gibco, Rockville, MD, USA) with 10% FBS (Gibco) and 1% penicillin–streptomycin (100 U/mL penicillin and 100 µg/mL streptomycin). For the best possible cell growth, the incubation temperature was 37 °C and the humidity level was 5% CO₂. The shRNAs that target KLHDC8A were manufactured by Jikai Gene in Shanghai, China. An online RNA interference (RNAi) design tool was used to validate the specificity and low off-target effects of the shRNAs, which were designed to target conserved areas of the KLHDC8A mRNA. Stable transfection and cell selection under puromycin selection pressure are made possible by cloning the shRNA sequences into the lentiviral vector pcDNA-EF2-puromycin. As a control for all tests, the same lentiviral vector was also used to clone a non-targeting scrambled shRNA gene. The Lipofectamine 2000 reagent (Thermo Fisher Scientific, Waltham, MA, USA) was used to transfect U251 and U87 cells with lentiviral vectors carrying KLHDC8A-specific shRNAs or scrambled control shRNAs, according to the manufacturer's procedure. After 48 h of transfection, the lentiviral particles were collected, filtered, and concentrated by ultracentrifugation.
2.1.4 RNA extraction and real-time PCR assay
The TRIzol reagent (Sigma-Aldrich, USA) was used to extract total RNA from astrocytes and glioma cell lines (TG905, LN308, T98, U87, U251) according to the manufacturer's instructions. To lyse the cells, 1 mL of TRIzol reagent was added per well after cell harvesting from the growth plates. Two hundred microliters of chloroform were added after homogenization, and the samples were spun at 12,000 times the force of gravity for fifteen minutes at 4 °C. The RNA-containing water phase was cautiously gathered, and 500 µL of isopropanol was added to precipitate the RNA. The RNA pellet was resuspended in water that did not contain RNase after being washed with 75% ethanol following centrifugation. PrimeScript RT Reagent Kit (TaKaRa, Japan) cDNA synthesis was performed according to the manufacturer's procedure. Twenty microliters of RNAse-free water were added to four and a half milliliters of 5 × PrimeScript buffer, one microliter of PrimeScript RT enzyme mix, one microliter of random hexamers, and one microliter of oligo(dT) primers. The enzyme was inactivated at 85 °C for 5 s after the reaction had been incubated at 37 °C for 15 min. The cDNA that was produced was then frozen at -20 °C until it could be used again in real-time PCR. An ABI 7500 Real-Time PCR equipment (Applied Biosystems, USA) was used to conduct real-time quantitative PCR (qPCR) with the SYBR Green Real-Time PCR Kit (TaKaRa, Japan). A 20 µL reaction volume was used for the reactions, which included 1 µL of cDNA template, 10 µL of 2 × SYBR Green PCR Master Mix, 0.4 µL of forward and reverse primers (10 µM each), and 8.2 µL of nuclease-free water. The comparative Ct (2^ − ΔΔCt) approach was used to do relative quantification of KLHDC8A expression. Fold changes in gene expression were calculated relative to the control (normal astrocyte cells), and Ct values for KLHDC8A were adjusted to GAPDH as an internal control. For determination of the expression of KLHDC8A, the following primers were used: forward, 5′-ATGGAGGTGCCTAACGTCAAG-3′, reverse 5′- CCGTTGTCGTCACATCCCC-3′; for GAPDH: forward 5′-GTCAAGGCTGAGAACGGGAA-3′, reverse 5′-AAATGAGCCCCAGCCTTCTC-3′.
2.2 Cell proliferation assay
In vitro cell proliferation tests were conducted using the U251 and U87 cell lines to assess the effect of KLHDC8A knockdown on glioma cell proliferation. Complete DMEM (supplemented with 10% FBS and 1% penicillin–streptomycin) was used to resuspend trypsinized U251 and U87 cells after 36 h of transfection. To guarantee uniform seeding density in all experimental wells, the cell density was calibrated to 3 × 104 cells per well using a hemocytometer. 100 µL of single-cell suspensions were added to each well of 96-well plates. Three independent platings were performed on each treatment group (shKLHDC8A and scrambled control) to guarantee statistical reliability. For as long as 96 h, the plates were kept in an incubator with a humidifier and 5% CO₂ at 37 °C. At 24, 48, 72, and 96 h after seeding, the CCK-8 reagent was used to evaluate cell proliferation at 24-h intervals. Each well was supplemented with 10 µL of CCK-8 solution at each time point, and the cells were left to incubate at 37 °C for an extra four hours. Each well's optical density (OD) was measured at 450 nm using a microplate reader (BioTek, USA) after the 4-h incubation with CCK-8. One way to measure cell proliferation over time is by looking at the optical density (OD) measurements, which are directly related to the number of viable cells. Proliferation curves for cells were created using the OD values measured at 24, 48, 72, and 96 h. Relative cell proliferation rates were presented as a percentage of the mean OD value at 24 h after triplicate wells were measured.
2.3 Colony formation assay
A colony formation experiment was conducted to evaluate the clonogenic potential and long-term proliferative capacity of U251 and U87 cells after KLHDC8A knockdown. Cells from U251 and U87 were transfected for 36 h, then trypsinized, hemocytometer counted, and resuspended in full DMEM (10% FBS). With a final amount of 2 mL DMEM, 500 cells per well were seeded into 6-well plates for both the shKLHDC8A and scrambled control groups. To make sure that each cell could grow into its own distinct colony, a low seeding density was used. For 10 days, the cells were kept at 37 °C in a humidified environment containing 5% CO₂. In order to keep the cells growing at their optimal rate, the medium was changed every three days. The cells were given permission to multiply and cluster into colonies, which were characterized as groups of 50 cells or more. Gently aspirating the medium and washing the cells twice with phosphate-buffered saline (PBS) to remove remaining media was done after 10 days of culture, when noticeable colonies had formed. The next step was to fix the cells at room temperature with 4% paraformaldehyde for 15 min. After the colonies were fixed, they were stained for 20 min with a 0.5% crystal violet solution (Sigma-Aldrich, USA) in order to see them. Because crystal violet attaches to DNA, counting and identifying colonies is a breeze. To remove any excess stain, the wells were delicately rinsed with distilled water until the background became clear and the stained colonies remained. At room temperature, the plates were let to dry naturally. We used a digital camera (Olympus Inc.) attached to a microscope to take pictures of the dyed colonies, and then we numbered them by hand.
2.4 Western blot assay
After 48 h of transfection, U251 and U87 GBM cells were analyzed for shRNAs targeting KLHDC8A or control shRNAs that were scrambled. After rinsing with cold PBS, cells were lysed on ice in RIPA buffer (Beyotime, China), which also contained protease and phosphatase inhibitors (Sigma-Aldrich). To guarantee full lysis, cell lysates were incubated on ice for 30 min while being sometimes vortexed. To remove cell debris, the lysates were spun at 12,000 × g for 15 min at 4 °C. Then, the liquid that remained after centrifugation, which included total proteins, was collected. We followed the manufacturer's instructions to determine the protein concentrations using a bicinchoninic acid (BCA) protein assay kit from Beyotime, China. The absorbance was measured at 562 nm using a microplate reader (BioTek, USA) after 10 µL of each protein sample was combined with BCA reagent. A standard curve was made using bovine serum albumin (BSA) standards to determine protein concentrations. The proteins were denatured by boiling 30 µg of protein per sample with 5 times the volume of loading buffer at 95 °C for 5 min. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) was used with a 10% polyacrylamide gel to separate the protein samples. Until the dye front reached the bottom of the gel, electrophoresis was performed at a constant voltage of 100 V for around 1.5 h. Using a wet transfer technique, the proteins were moved from the gel onto a PVDF membrane (Millipore, USA) after separation. The transfer was carried out in a transfer buffer containing 25% methanol, 25% tris, and 192% glycine at 4 °C for 1 h at 100 V. A short wash in Tris-buffered saline with Tween-20 (TBST) and blocking were performed on the PVDF membrane following transfer. After preparing 5% skim milk in TBST, the PVDF membrane was blocked for 1 h at room temperature with careful shaking to avoid non-specific antibody binding. To get rid of any leftover blocking solution, the membrane was rinsed three times with TBST. Primary antibodies specific to the following proteins were added to the membrane and left to incubate overnight at 4 °C. These antibodies were from Cell Signaling Technology and were used at a ratio of 1:1,000 for β-catenin, CDK4 and CDK6, respectively. GAPDH was added at a ratio of 1:5,000 from Beyotime. To achieve the highest level of binding specificity, the antibodies were diluted in TBST that included 1% BSA. The membrane was washed three times with TBST, with each wash lasting 10 min, after being incubated overnight. One hour of incubation at room temperature followed the primary antibody incubation, and then the membrane was treated with secondary antibodies conjugated with horseradish peroxidase (HRP) (anti-rabbit IgG, 1:1,000; Beyotime, China). To eliminate any unbound antibodies, the membrane was washed three times with TBST, with each wash lasting 10 min, after the secondary antibody incubation. A Millipore, USA-based enhanced chemiluminescence (ECL) reagent was used to identify the bound antibodies. The Amersham Imager 600 (GE Healthcare, USA) was used to visualize the signal after the membrane was incubated with the ECL solution for 1 min. The strength of the bands dictated the adjustment of exposure periods, which were then used to collect and preserve photos for analysis. To conserve valuable samples, we used partial membranes (not full-length) in our experiments and cropped the membranes to focus on the target protein region.
2.5 Statistical analysis
The means ± SDs are used to express the study measurements, and each assay was conducted three times. We used Student's t test to find statistical differences between two groups and one-way ANOVA to find statistical differences among several groups. When comparing multiple groups, we first ran a one-way ANOVA and then used Tukey's post hoc test to look at how the groups stacked up against each other when comparing them in pairs. In order to determine if there was a statistically significant difference between the group means, we used a one-way analysis of variance (ANOVA) that controlled for type I error and multiple comparisons. An area under the curve (AUC) was computed in ROC curve analysis to evaluate the accuracy of KLHDC8A expression in predicting patient outcomes. The AUC values were used to display the calculated sensitivity and specificity as well. The statistical studies were carried out using the following software: R (v.4.0.3), RStudio (v.1.3.1093), SPSS version 20.0, GraphPad Prism 8.0, And Microsoft Excel 2016. To be deemed statistically significant, a P value had to be lower than 0.05.





留言 (0)