Mice
In vivo experiments were performed using 8-week-old female BALB/cOlaHsd mice purchased from Envigo (Amsterdam, the Netherlands). Animal procedures were performed in accordance with the Swiss Animal Act (455.109.1—September 2008, 5th) at the DBMR of the University of Bern. All animal experiments were conducted by protocols approved by the Cantonal Veterinary Office Bern, Switzerland (License: BE80/2021).
To generate sporozoites, animal experiments were conducted in strict accordance with the guidelines of the Swiss Tierschutzgesetz (TSchG; Animal Rights Laws) at the Institute of Cell Biology and approved by the ethical committee of the University of Bern (Permit Number: BE98/19 and BE118/2022). Mice were kept in specific pathogen-free conditions and subjected to regular pathogen monitoring by sentinel screening. Moreover, mice were kept in individually ventilated cages furnished with autoclaved aspen woodchip, a mouse house, and paper tissue at 21 ± 2 °C under a 12:12 h light–dark cycle at a relative humidity of 55 ± 10%. Animals were fed with a commercially prepared autoclaved dry rodent diet and water, both available ad libitum. The health of animals was monitored daily by visual health checks and the parasitemia of infected animals was determined by flow cytometry.
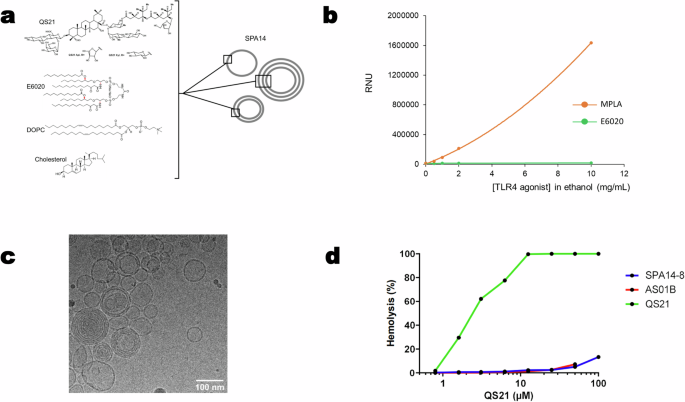
Production and purification of vaccine candidates
Manufacturing of CuMVTT control was described in detail in Zeltins et al.46. VLP-based malaria vaccine candidates were produced as mosaic particles consistent of unmodified CuMVTT and modified CuMVTT subunits genetically fused to sequences of the PfCSP, similar as described for VLP Peanut58.
Briefly, the CMV-Ntt830 gene was inserted under the T7 promotor into the pET Duet-1 vector (Novagen, Germany, Cat. 71146). The product was amplified by PCR and cloned into the pTZ57R/T vector using the InsTAclone PCR Cloning Kit (Fermentas, Lithuania; Cat. K1214) in E. coli XL1-Blue cells (Agilent, USA, Cat. 200249). Correct plasmid clone formation was verified using the BigDye Cycle Sequencing Kit (Thermo Fisher Scientific, USA; Cat. 4337449) as well as the ABI Prism 3100 Genetic Analyzer (Applied Biosystems, USA). Plasmids were treated with HindII restriction enzyme (Thermo Fisher Scientific, USA; Cat. ER0502), Klenow enzyme (Thermo Fisher Scientific, USA; Cat. EP0422) Klenow enzyme as well as the Ndel restrictase (Thermo Fisher Scientific, USA; Cat. ER0581) and obtained fragment sub-cloned into the Ndel/EcoRV sites of the pET Duet-1 vector (Novagen, Germany, Cat. 71146) resulting in pETDu-CMV-Ntt830. The CMVB3d-PfCSP gene was excision from pACYCDu-CMVB3d-PfCSP (pACYCDu: Novagen, Germany, Cat. 71147) using NcoI (Thermo Fisher Scientific, USA; Cat. ER0572) and HindIII (Thermo Fisher Scientific, USA; Cat. ER0502) enzymes and inserted under the T7 promotor of pETDu-CMV-Ntt830. Restriction enzyme analysis was performed to identify correct clones (pETDu-CMVB3d-PfCSP-CMVtt830).
E. coli C2566 (New England Biolabs, USA) cells were transformed with the plasmid pETDu-CMVB3d-PfCSP-CMVtt830. Successfully transformed E. coli cells were grown in 2TY medium (Trypton 1.6%, yeast extract 1%, 0.5% NaCl, 0.1% glucose) containing ampicillin (50 mg/L) on a rotary shaker (10 rpm, 30 °C) to OD600 absorption of 0.8. Cultures were induced with 0.2 mM IPTG and the medium was supplemented with 5 mM MgCl2. Incubation was continued on the rotary shaker (10 rpm, 20 °C, 18 h), and resulting biomass was collected by centrifugation (15,000 × g, 4 °C, 10 min).
Cells were disrupted by sonication for 16 min on ice (Hielscher 200, power 70%, pulse 50%) in the following buffer: 20 mM Tris, 5 mM EDTA, 5 mM Et-SH, 5% glycerol, 10% sucrose (pH 8.0); 0.5% TX-100 was added and solution was rotated (10 rpm) overnight at 4 °C.
The solution was centrifuged for 10 min at 10,000 rpm (Eppendorf 5804) and soluble fraction was loaded on a 20–60% sucrose gradient in the following buffer: 20 mM Tris, 2 mM EDTA, 5% glycerol, 0.5% TX-100. Centrifugation was performed for 6 h at 18 °C at a speed of 25,500 rpm (Beckman Coulter SW32). Fractions were analyzed by SDS–PAGE and VLP containing fractions sedimented by centrifugation (50,000 rpm, 4 h, 4 °C; Beckman Coulter 70Ti). Pellet was dissolved in 20 mM Tris, 2 mM EDTA (pH 8.0) and loaded on a 30% sucrose cushion containing 20 mM Tris, 2 mM EDTA, 5% glycerol, 0.5% TX-100 (72,000 rpm, 1 h, 4 °C; Beckman Coulter TLA-100.3). Obtained VLPs were solubilized in (20 mM TRIS, 2 mM EDTA, pH 8.0) and analyzed by SDS–PAGE, agarose gel, TEM, and DLS (Figs. 1 and 3). Endotoxin levels for all vaccine candidates were below 100 EU per injection.
SDS–PAGE analysis
15 µL sample (1 mg/mL) was mixed with 3 µL reducing buffer (Thermo Scientific, Cat. 39000), heated for 5 min at 95 °C and then loaded onto a 12% SDS–PAGE with a 4% stacking gel. 6 µL protein ladder (Thermo Scientific, Cat. 26616) were loaded and the gel was run at 70 V in the following buffer: Tris (hydroxymethyl)-aminoethane 2.5 mM, glycine 25 mM, SDS 0.01%. Protein bands were stained with InstantBlue® Coomassie Protein Stain (Abcam, Cat. Ab119211) and a gel picture was taken with Azure Biosystems c300 using the visible channel, auto-exposure time. Incorporation rate of PfCSP-CuMVTT fused subunits into VLPs was determined as previously described22. Briefly, protein band intensities were quantified with FIJI Image J software v1.53c84. The PfCSP-CuMVTT-derived protein band intensity was divided by the corresponding CuMVTT-derived protein band intensity for each sample. The obtained ratio was corrected for the differed molecular masses of the proteins and multiplied by 180, the total number of subunits of CuMVTT46.
Agarose gel
15 µL sample (1 mg/mL) was mixed with 2.5 µL loading dye (New England BioLabs, Cat. B7024S) and loaded with ladder (Thermo Scientific, Cat. SM0242) onto a 2% agarose (BioConcept, 7-01P02-R) gel, run in Tris–borate–EDTA buffer at 50 V. Gel picture was taken with Azure Biosystems c300 using the UV302 channel, auto-exposure time.
Transmission electron microscopy (TEM)
VLP formation was confirmed for each vaccine sample produced by TEM (Philips CM12 EM). For this purpose, sample grids were glow discharged and 10 μL of VLP sample (1 mg/mL) was added for 30 s. Afterward, Grids were washed three times with ddH2O and negatively stained with 5 μL of 5% uranyl acetate for 30 s. Excess of uranyl acetate was removed by pipetting. Grids were air-dried for 10 min and images were taken with 84,000× and 110,000× magnification.
Dynamic light scattering (DLS)
Average VLP particle size in vaccine batches was measured by DLS using the Zetasizer Nano ZS instrument (Malvern Panalytical Ltd., UK), and measurements were evaluated with DTS software v. 6.32 (Malvern Panalytical Ltd., UK). 3 consecutive measurements were performed per sample.
Vaccination regimen for naïve mice
Mice were immunized subcutaneously (s.c.) with 30 µg vaccine candidate or CuMVTT control in 100 µL PBS using an insulin syringe (B Braun Omnican®, Cat. 9151133S). 28 days post prime a booster injection with an equal dose was given. Serum samples were collected the day of prime (D0), the day of boost (D28, before vaccination), and 10 days post boost (D38, before sporozoite challenge) via tail-vein bleeding, and serum was isolated using BD Microtainer® Collection Tube (BD Biosciences, Cat. 365967).
Parasite lines
For the challenge and IFA experiments a chimeric Pb parasite line in which the endogenous CSP gene was replaced with the Pf counterpart has been used. This chimeric parasite line was generated by Triller et al. and obtained from the Rodent Malaria genetically modified Parasites database (RMgmDB; ; 2257 cl2, RMgm 4110)31. Brief description of the parasite line: the endogenous Pb CSP was replaced with the Pf CSP in the parental line (676 cl1, RMgm-29; Janse et al.85). This parental line expresses a fusion protein of GFP (gfp-mu3) and Luciferase Firefly (LucIAV) under the expression of the eef1a promoter at the 230p silent locus85. The resulting line, Pb/PfCSP-GFP-Luc, has been referred to as Pb/PfCSP in our study. Importantly, these chimeric Pb/PfCSP parasites exhibited robust sporozoite development in mosquitoes, with successful expression of PfCSP on the sporozoite surface. Furthermore, the in vivo infectivity of Pb/PfCSP parasites in wild-type mice resembled that of parasites from the parental Pb line31,86,87. For IFA, the parental line (676 cl1, RMgm-29; Janse et al.85) has been used as a control.
Mosquito, parasite maintenance, and salivary gland sporozoites dissectionMosquito maintenance
Anopheles stephensi (A. stephensi) mosquitoes were bred at 28 °C, 80% humidity, and a 12 h light–dark cycle in an incubator (MLR-352H SANYO, Japan) in the insectarium at the Institute of Cell Biology of the University of Bern. Mosquitoes were daily fed with cotton pads containing 8% fructose solution supplemented with 0.2% PABA (Sigma-Aldrich, Cat. No. 100536) and female mosquitoes were fed with human blood once a week to allow them to lay eggs. The next day, eggs were collected, washed once with 70% ethanol, and twice with water before being added to a metal bowl with a drop of about 10 µl of NobilFluid Artemia (JBL 33801 Germany), where they developed into larvae. After 2 days in the metal bowl, the larvae were transferred into a water-containing plastic bowl and fed with grounded Tetra TabiMin complete food tablets (Olibetta, 400080, Germany). After 7 days, pupae were collected and put in a stock cage, where they were kept at 27 °C and 80% humidity until hatching. Adult mosquitoes were fed with 8% fructose supplemented with 0.2% PABA (Sigma-Aldrich, Cat. No. 100536). After 2 days, female A. stephensi mosquitoes were ready to be infected with Pb/PfCSP parasites and could therefore be collected from the stock cage. After feeding, mosquitoes were kept at 20 °C.
Parasite maintenance
Female BALB/cOlaHsd mice (6–8 weeks; Janvier Laboratories, France) were used to maintain parasites and for mosquito feeding with parasites. Mice were injected with Pb-infected blood via an intraperitoneal route and when parasitemia reached 2%–5%, mice were euthanized in a CO2 chamber and parasites isolated following exsanguination and the infected blood was intravenously passaged into a naïve mouse for further mosquito feeding. Upon reaching a parasitemia of 5%–7% and verifying for gametocytes presence (0.5%–1%), mice were anesthetized with a terminal dose of ketamine: xylazine, and when no longer reacting to touch stimulus was placed on a cage of ~100–150 mosquitoes for 45 min at 20 °C. Infected mosquitoes were kept at 20 °C and 80% humidity and fed with 8% fructose, containing 0.2% PABA for 16 and up to 26 days for salivary gland sporozoite dissection.
Salivary gland sporozoites dissection
Mosquitoes were aspired with a vacuum (Fulton U.S. MX-991/U, Hausherrs Machine Works, USA), anesthetized with chloroform and stored during dissection on ice. Before dissection, mosquitoes were dipped into PBS, 70% ethanol, and again into PBS to be placed on a slide, and salivary glands were extracted under the binocular (SZX 10 Olympus, Japan) with forceps and collected in a tube containing 50 μL of non-complement Iscove’s Modified Dulbecco’s Medium (IMDM). Salivary glands were homogenized (Pellet pestles Cordless Motor, Sigma-Aldrich, USA) and counted with a Neubauer counting chamber (VWR International GmbH, Switzerland) before being mixed with PBS and i.d. injected into mice.
Challenge of vaccinated mice with Pb/PfCSP sporozoites
Previously immunized mice were challenged with Pb/PfCSP sporozoites 10 days after the booster injection. To this end, mice were inoculated with 5000 sporozoites given in 20 µL PBS i.d. (10 µL into each ear) by using an insulin syringe (B Braun Omnican®, Cat. 9151133S). Parasitemia was monitored daily by flow cytometry. For this purpose, 1 µL blood from the tail vein was mixed with 500 µL PBS supplemented with 2% FBS and 100 mM EDTA. Pb/PfCSP-infected erythrocytes of blood samples were analyzed by flow cytometry (CytoFLEX, Beckman Coulter) for GFP positivity (Pb/PfCSP parasites express GFP as described above). Data were collected using CytExpert™ Software, version 2.6. Data were analyzed by using FlowJo™ Software, version v10.7 (BD Biosciences). Mice having a measured parasitemia >0.1% were considered infected. Mice having a measured parasitemia >2% were euthanized (termination criterion).
Production and purification of recombinant PfCSP (rPfCSP)
The production of recombinant PfCSP followed the procedure previously described41. Briefly, PfCSP sequence of the Pf3D7 strain (PlasmoDB ID: PF3D7_0304600.1) was codon-optimized for the gene expression in mammalian cells (GenScript, USA). The first 20 leader amino acids were changed to a mammalian secretory signal peptide derived from the modified bovine lactine (MDSKGSSQ KGSRLLLLLVVSNLLLPQGVLA) and the GPI-anchor residues 376–397 were replaced with a GSG-linker followed by an 8x-histidine-tag. The gene construct was cloned into a pcDNA3.1(+) expression vector under the control of the T7 promotor (GenScript, USA). rPfCSP was expressed by using the Expi293™ expression kit (Thermo Scientific, Cat. A14635) according to the manufacturer’s protocol. The supernatant was harvested 5 days post-transfection and dialyzed to PBS using Spectra/Por™ dialysis membrane (Fisher Scientific, Cat. 08-667B). Protein purification was carried out by affinity chromatography using His-Trap HP columns (Cytiva, Cat. 17524701) and the Äkta go™ (Cytiva, USA) purification system. Protein concentration was determined using Pierce™ BCA Protein Assay Kit (Thermo Scientific, Cat. 23225). Correct expression was verified by SDS–PAGE (previously described here) and western blot analysis. For western blot, rPfCSP was separated on SDS–PAGE, and protein bands were transferred onto a 0.2 µM nitrocellulose membrane (BioRad, Cat. 1704158) using the Trans-Blot® Turbo™ Transfer System (BioRad, USA) and further processed by using the iBind™ Flex Western Device (Invitrogen, USA) according to the manufacturer’s instructions. His-tag-carrying protein bands were detected with the HRP anti-His Tag antibody (BioLegend, Cat. 652504), 1:1000 diluted. PfCSP protein bands were detected with the anti-PfCSP antibody (obtained through BEI Resources, NIAID, NIH: Monoclonal Anti-Pf CSP, Clone 2A10 (produced in vitro), MRA-183A, contributed by Elizabeth Nardin) used as primary antibody at 1:2000 dilution and HRP anti-mouse IgG (Jackson ImmunoResearch, Cat. 115-035-071) at 1:1000 dilution as secondary antibody. Membrane was developed by using SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo Scientific, Cat. 34579) and the image was obtained with Azure Biosystem c300, 5 s exposure time.
Enzyme-linked immunosorbent assay (ELISA)
Pb/PfCSP sporozoite and rPfCSP-specific IgG antibodies in serum of immunized mice were detected by ELISA. For this, ELISA plates (CORNING, Cat. 3690) were coated with 50 µL Pb/PfCSP sporozoites diluted in PBS (30 sporozoites/µL) or 50 µL rPfCSP diluted in PBS (1 µg/mL) overnight at 4 °C. ELISA plates were washed four times with PBS using the Microplate washer (BioTek 405 TS Microplate Washer, Agilent), 100 μL/well PBS. Unspecific binding was prevented by treating plates with PBS-Casein 0.15% for 2 h at room temperature (100 μL/well). Plates were flicked and mouse serum 1:20 pre-diluted in PBS-Casein 0.15% was added to the top row of the plate. A 1:3 serial dilution in PBS-Casein 0.15% was performed and plates were incubated for 1.5 h at room temperature (total volume of 50 µL/well). Subsequently, plates were washed with PBS-0.01% Tween20 for four times with Microplate washer (100 μL/well), and HRP goat anti-mouse IgG (Jackson ImmunoResearch, Cat. 115-035-071) was added at 1:1000 dilution in PBS-Casein 0.15% (50 µL/well), plates were incubated for 1 h at room temperature. Plates were washed with PBS-0.01% Tween20 for four times with Microplate washer (100 μL/well) and developing solution (30 mM citrate buffer including 5% tetramethylbenzidine and 1.5‰ H2O2) was added (50 μL/well). The color reaction was stopped by the addition of 1 M H2SO4 (50 μL/well) and absorbance at 450 nm (OD450) was read with BioTek Cytation 5 imaging reader. Half-maximal antibody titers (OD50) were defined as the reciprocal of the dilution leading to half of the OD measured at saturation.
rPfCSP-specific IgG subclass and IgM antibodies were determined following the same protocol but using a different detection antibody: HRP rat anti-mouse IgG1 (BD Biosciences, Cat. 559626, 1:1000 dilution), HRP rat anti-mouse IgG2a (BD Biosciences, Cat. 553391, 1:1000 dilution), HRP goat anti-mouse IgG2b (Invitrogen, Cat. M32407, 1:1000 dilution), HRP goat anti-mouse IgG3 (Southern BioTech, Cat. 1101-05, 1:1000 dilution), HRP goat anti-mouse IgM (Jackson ImmunoResearch, Cat. 115-035-075, 1:1000 dilution).
High-avidity IgG antibodies specific for rPfCSP were determined by a previously described protocol63. For this, the normal ELISA protocol was extended by an additional washing step. Two similarly coated plates incubated with the same sera were washed upon sera incubation either three times for 5 min with PBS-0.01%Tween20 or three times for 5 min with 7 M urea in PBS-0.01% Tween20 (50 µL/well). Afterwards, detection antibody was added and the above-described protocol continued. The AI was calculated by AIx = (OD450 (dilution ×) urea treated plate)/(OD450 (dilution ×) non-urea treated plate).
Epitope mapping
To map the epitopes of IgG antibodies induced by the different malaria vaccine candidates, an ELISA-based method was employed. ELISA plates (CORNING, Cat. 3690) were coated with 50 µL of BSA-conjugated peptides diluted in PBS (5 µg/mL) and incubated overnight at 4 °C. The peptides used were as follows: H-CKHKKLKQP-NH2, H-CDGNPDPNA-NH2, H-CNANPNVDP-NH2, and H-CNANPNANP-NH2, all conjugated to BSA via a cysteine residue and purchased from BIOSYNTH®. Following peptide coating, the standard ELISA protocol was carried out as previously described.
Immunofluorescence assay (IFA)
Salivary gland sporozoites were dissected as previously described. Collected sporozoites were resuspended in minimum essential medium (MEM; BioConcept, 1-31F01-l) supplemented with 1% l-glutamine (Bioconcept, 5-10K00-H) and 1% penicillin–streptomycin (Bioconcept, 4-01F00-H) and complemented with 10% heat-inactivated fetal calf serum (FCS, GE Healthcare) before being deposited (300 µL) on cover slides and placed in a 24-well plate. The 24-well plate was centrifugated for 1 min at 1000 rpm at RT before the plate was incubated for 2 h at 37 °C allowing sporozoite to be deposited and glide on the slide. Thereafter the remaining medium was carefully aspirated and sporozoites were fixed with 4% PFA in PBS for 20 min at RT. After three washing steps with PBS, cells were permeabilized with 0.15% Triton X-100 in PBS for 10 min. After another three washing steps with PBS, unspecific binding was blocked by incubating cells with 3% BSA in PBS for 1 h at RT. Mouse sera diluted 1:1000 in BSA 3% in PBS and primary antibodies: (1) anti-PbTRAP rabbit (kindly provided by Prof. Freddy Frischknecht, diluted 1:500 in BSA 3% in PBS); (2) anti-PbCSP rabbit (Eurogentec, diluted 1:1000 in BSA 3% in PBS); (3) anti-PfCSP (obtained through BEI Resources, NIAID, NIH: Monoclonal Anti-PfCSP, Clone 2A10 (produced in vitro), MRA-183A, contributed by Elizabeth Nardin, diluted 1:1000 in BSA 3% in PBS) were given on the cover slides and these then incubated for 1 h at RT. After washing three times with PBS, fluorescently labeled secondary antibodies diluted in BSA 3% in PBS were added: (1) anti-rabbit Cy5 (Dianova; 1:2000); (2) anti-mouse Alexa488 (Invitrogen A-11001, 1:2000); (3) anti-mouse Alexa594 (Invitrogen A-11032 1:2000), and the samples were incubated for 1 h at RT protected from light. Washing three times with PBS was followed by a 10-min incubation with DAPI (Sigma, D9542, 1 μg/mL) for 10 min. Cover slides were then washed three times with PBS and mounted onto microscopy slides using Dako Fluorescence Mounting Medium. Stained sporozoites were imaged on a Leica TCS SP8 laser-scanning confocal microscope. Acquired images were processed with FIJI software.
In the control staining named “no serum”, no mice sera were added to sporozoites, but only secondary antibodies to control for the unspecific binding of these secondary antibodies to sporozoites.
Statistics
Data are presented as mean ± SEM and were statistically analyzed using ordinary one-way ANOVA with Tukey correction for multiple comparisons or Log-rank (Mantel–Cox) test for survival curves. Pearson correlation coefficient was calculated to assess linear relationship between the number of targeted NANP repeats and the induction of rPfCSP-specific high-avidity IgG. A saturation curve fit was applied using nonlinear regression to model induction of rPfCSP-specific IgG/IgM as a function of targeted NANP repeats. Analyses were performed using GraphPad PRISM 10.3 (GraphPad Software Inc., USA). The value of p < 0.05 was considered statistically significant. Statistical significance is noted in figures as *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.


留言 (0)