
記住我

The primary aim of this study was to evaluate the effectiveness (infection cure rate) of preformulated Bactisure® irrigation solution in vivo compared with saline solution in a control group in cases of acute or haematogenous knee or hip periprosthetic infections treated with DAIR.
Design and settingThis study is a prospective single-centre randomized controlled trial involving patients with acute haematogenous PJI who receive standard DAIR surgery from December 2022 to December 2024. The type of irrigation solution used during the surgery will define the two groups allocated at a 1:1 ratio: a control group with saline solution and an experimental group receiving the Bactisure® preformulated solution. This interventional study will be single-blinded, with the participants blinded for group allocation.
This study will be under the oversight of the Vall d’Hebron Institut de Recerca (VHIR) in Barcelona, Spain. While the institution itself will not directly participate in the study design, data collection, analysis, management, interpretation, or dissemination of results, it provides a supportive environment for the study’s execution. The day-to-day operations and coordination of the trial will be managed by the faculty members of the Septic and Reconstructive Surgery Unit from the Orthopaedic Surgery Department at Vall d’Hebron University Hospital. This department is responsible for trial oversight, ensuring adherence to the study protocol, and conducting regular meetings to review progress.
The trial was set up according to CONSORT guidelines [20], and we also used the SPIRIT checklist when writing our report [21]. Ethical approval has already been acquired by Vall d’Hebron University Hospital Clinical Research Ethics Committee (approval number: PR(AT)192/2022). Additionally, the patients’ written consent will be obtained before they participate in the study. The study is listed with the International Standard Randomized Controlled Trial Number (ISRCTN): https://doi.org/https://doi.org/10.1186/ISRCTN10873696 (registered on 19/12/2023).
In the event that protocol modifications are necessary after the trial has commenced, a request for review will be submitted to the Ethics Committee. Any modification that could significantly impact participant welfare will require obtaining new informed consent and updating the revised protocol in the trial registry.
ParticipantsThe inclusion and exclusion criteria are shown in Table 2.
Table 2 The inclusion and exclusion criteria of the subjectsPatients meeting the inclusion criteria will be presented by investigators with the option to participate in the clinical trial. They will be adequately informed via the patient information sheet (Annex 1), and patients who wish to participate in the clinical trial must sign the informed consent form (Annex 2).
InterventionsAll patients with acute or haematogenous PJI will undergo standard surgical treatment, debridement, antibiotic, and implant retention (DAIR), with the type of irrigation solution used during surgery depending on the assigned group: saline solution (group 1) or preformulated Bactisure solution (group 2). During the DAIR surgery, irrigation will use a low-pressure pulsatile lavage system with the assigned irrigation solution at preintervention randomization:
Group 1: 3 L (L) of saline solution, followed by 3 L of saline solution and then another 3 L of saline solution (9 L in total).
Group 2: 3 L of saline solution, followed by 1 L of Bactisure®, and then another 3 L of saline solution (7 L in total).
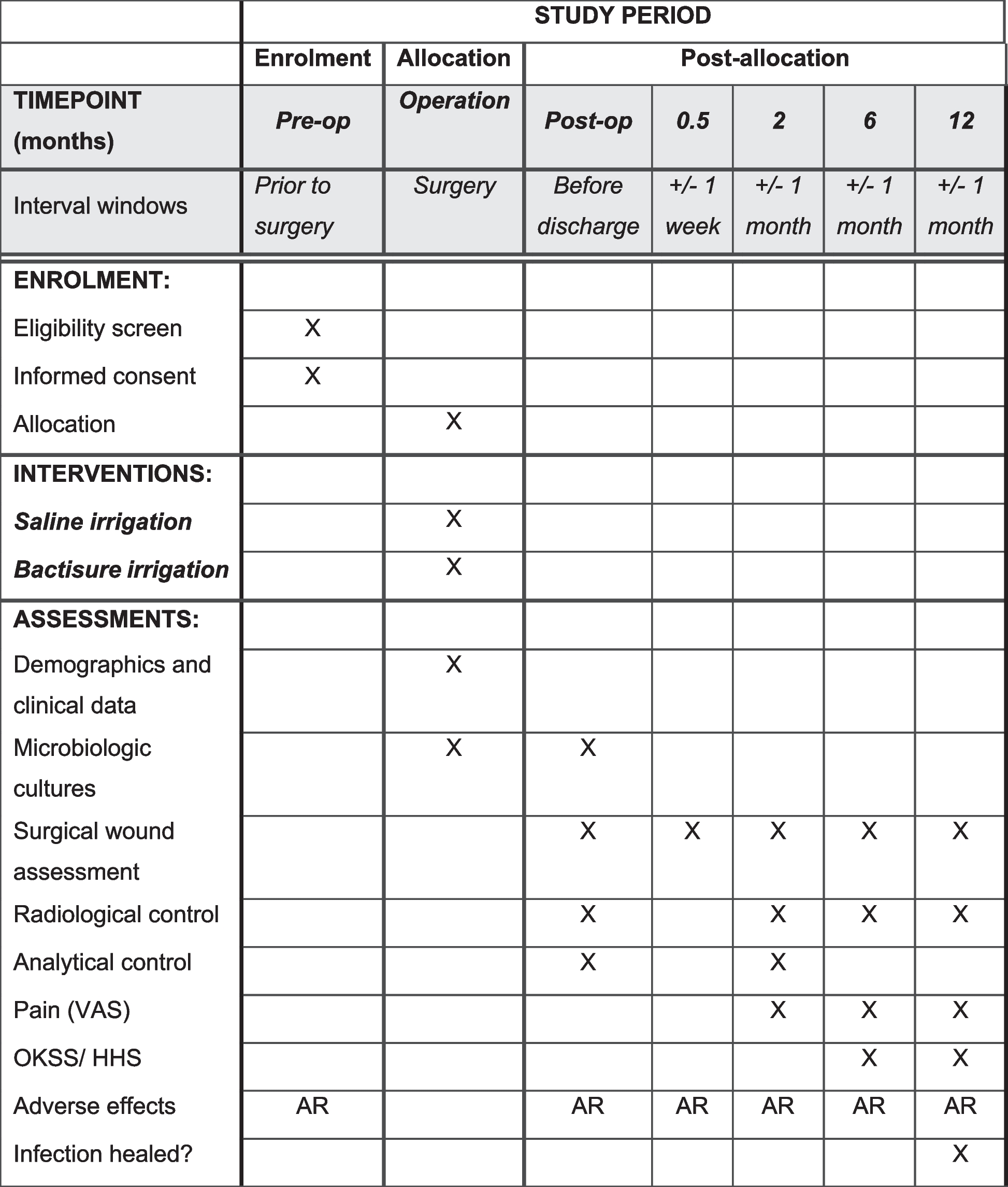
The necessary variables for the clinical trial will subsequently be collected throughout the clinical follow-up, which will last up to 12 months post-surgery. The detailed schedule of these collections is presented in Fig. 1, while the flowchart illustrating the trial’s participant progression is depicted in Fig. 2.
Fig. 1
Content for the schedule of enrolment, interventions, and assessments (SPIRIT 2013). AR as required
Fig. 2
Flowchart depicting patient selection
Data collection will be conducted via the SAP software program (SAP SE, Germany) in an anonymized Microsoft Excel online database. To safeguard the data, regular backups will be scheduled every quarter and stored securely in the cloud to ensure data integrity and availability.
Participant identification will be managed using unique numerical codes to ensure privacy, with all identifying information and consent forms securely stored in separate, restricted-access locations. All trial investigators are required to sign confidentiality agreements to ensure the protection of participant information throughout the study. Patient data will remain strictly confidential and will not be disclosed in any published research outputs.
One year after surgery, data collection for this clinical trial will cease, although routine follow-ups will continue within the respective healthcare units.
OutcomesThe primary outcome will be the infection cure rate measured via the following criteria at the 12-month follow-up. A case is considered cured if it meets all of the following requirements:
The absence of local recurrence, indicated by the absence of dehiscence, exudation, or fistula.
Radiographic evidence of no osteolysis.
There is no ongoing suppressive antibiotic treatment.
Absence of any reinterventions.
No deaths related to periprosthetic infection occurred.
The secondary outcome measurements will be as follows:
1.Length of hospital stay, in days, measured via medical records at one timepoint.
2.Potential postoperative complications (e.g. infections, reoperations, osteolysis) at hospitalization or in ambulatory controls measured via medical records at one timepoint.
3.Microorganisms identified on perioperative cultures at hospitalization were measured via standard laboratory methods at one timepoint.
Sample sizeTo calculate the required sample size (‘n’), we used the online software GRANMO (version 7.12, April 2012) from the Hospital del Mar Medical Research Institute (IMIM). Assuming a type α error of 0.05 and a type β error of 0.20, providing a statistical power of 80% in a bilateral test, 50 subjects (25 in each group) are required to detect a statistically significant difference between two proportions (reinfection rate). An expected reinfection rate of 0.45 for the control group (group 1) and 0.1 for the experimental group (group 2) was estimated. A follow-up loss rate of 5% was assumed. The sample size calculation is based on reinfection rates drawn from published literature, which have been adapted to meet the specific requirements of our clinical trial, assuming a reduction from 45% in the control group to 10% in the experimental group.
RandomizationThis single-centre randomized controlled trial comprises two groups allocated at a 1:1 ratio. To ensure balanced patient distribution, a restrictive randomization method using a computer-generated sequence will be employed, whereby cases are paired for randomization. The first case is assigned to either group 1 or group 2, and the second case is then placed in the opposite group. This process is repeated for each new pair of cases until the required sample size is reached.
BlindingA single-blind protocol will be employed throughout the trial, ensuring that patients remain uninformed regarding the specific irrigation solution utilized during their debridement procedures until the study’s completion. In contrast, surgeons will possess full knowledge of the irrigation solution being used; this is due to the distinct visual cues present in the packaging of the different solutions. Furthermore, the Bactisure® irrigation solution releases a distinctive, robust scent as a result of its acetic acid composition, which may be noticeable during its application. To minimize bias as much as possible, we have determined that an external investigator, blinded to group allocation, will conduct the analysis of the results.
Data analysis and statistical testsUpon completion of the clinical study, data will be analysed using R software, version 4.10 (R Foundation for Statistical Computing, Vienna, Austria). Quantitative variables will be compared via the Kruskal–Wallis exact test, whereas qualitative variables will be analysed via the chi-square test. A significance level of p < 0.05 will be applied to identify statistically significant differences. Univariate analyses will also be performed to identify potential risk factors for treatment failure. For variables showing statistical significance, a multivariate analysis will be conducted, employing logistic regression for qualitative variables. The primary analysis will be conducted using the intention-to-treat population. To compare treatment success and failure, both a time-to-event (survival) analysis and a final follow-up analysis will be performed. This approach allows us to capture both the timing of events and the overall outcome at the study’s conclusion.
An interim safety analysis will be performed upon completing interventions for the first 12 patients in each group. This analysis will assess adverse effects and the provisional effectiveness of both treatments.
Dissemination plansA variety of dissemination channels will be utilized to share information about the project and the study’s results with clinicians, patients, and the general public, including the following:
The study protocol will be published in a peer-reviewed, open-access journal before the recruitment phase concludes.
Upon completion of the study, the trial’s findings will be presented at both national and international meetings organized by orthopaedic surgeon-focused organizations. This will include events hosted by organizations such as the Spanish Society for Orthopaedics and Traumatology (SECOT) and the European Bone and Joint Infection Society (EBJIS).
The study report will be published in high-impact, peer-reviewed medical and orthopaedic journals with broad readership.
留言 (0)