
記住我

This is a prospective clinical trial designed as a single-center, open-label, randomized controlled study with enrolled patients allocated 1:1 and conducted at the First Affiliated Hospital of Shantou University Medical College, Shantou City, Guangdong Province, China. This protocol was designed in accordance with the Standard Protocol Project: Recommendations for Interventional Trials (SPIRIT) 2013 statement.
Study objectivesPrimary objective: the primary objective is to evaluate new tricuspid regurgitation or exacerbation of existing tricuspid regurgitation within 1 year of follow-up.
Secondary objective: the secondary objective is to evaluate the incidence of adverse cardiovascular events (acute heart failure, malignant arrhythmia, cardiogenic shock) or if pacemaker implantation surgery is required again.
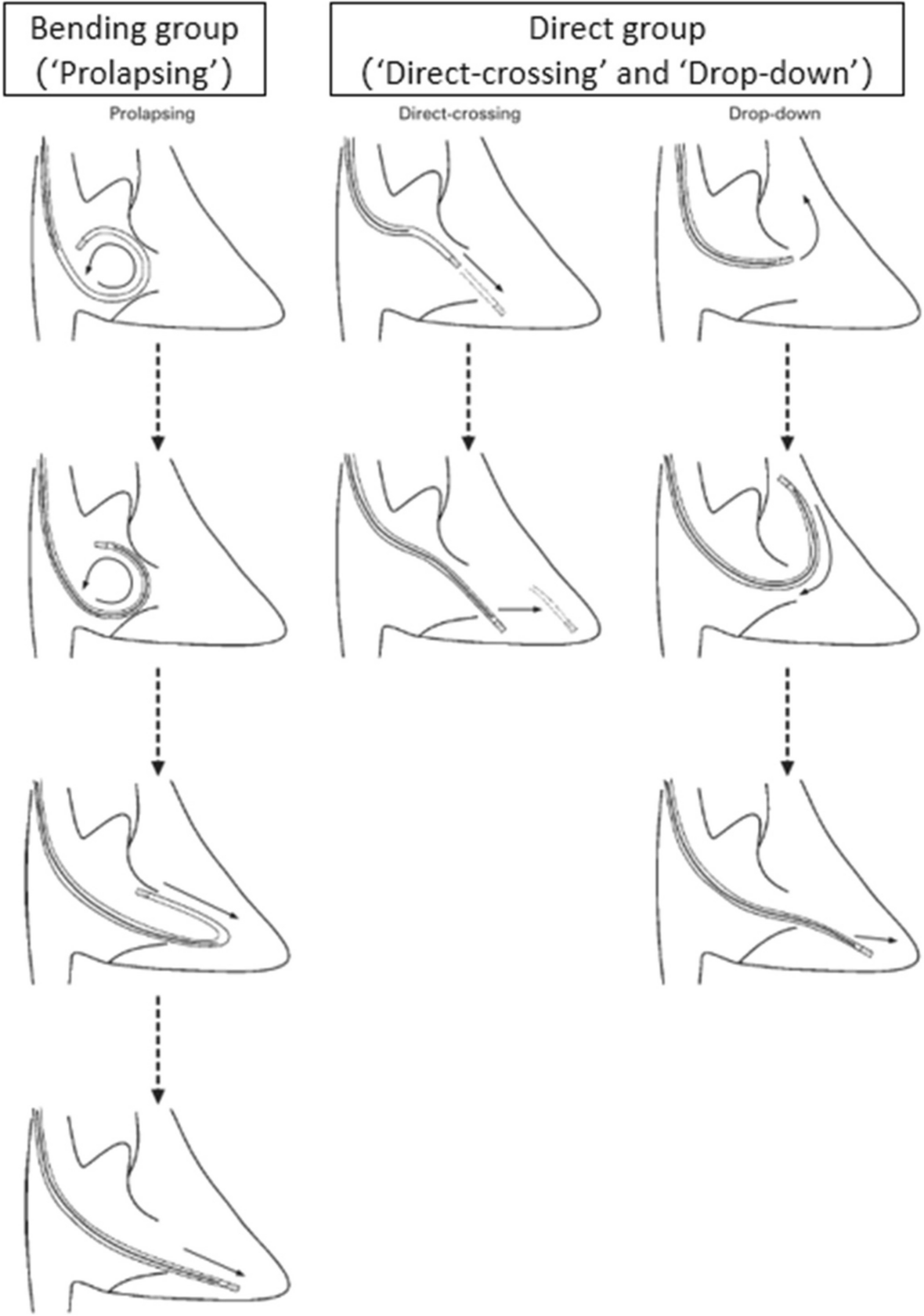
HypothesisSince the body of the curved lead is relatively round and blunt, we speculate that bending the pacing lead may more likely avoid penetrating the valve leaflet or intertwining with the chordae tendineae, and enter the right ventricle across the middle of the tricuspid annulus. The leeway of the lead is large, so that the valve leaflets and chordae tendineae are not easily restricted by the lead, thereby reducing the incidence of LITR and adverse cardiovascular events.
Ethics issuesThe present study protocol was approved by the Committee of Ethics of the First Affiliated Hospital of Shantou University Medical College (Number: B-2021–022).
Participant recruitmentClinical registration for this study was completed on April 19, 2021. After registration, patients who underwent permanent pacemaker implantation in the First Affiliated Hospital of Shantou University Medical College (a tertiary care hospital with independent interventional procedures for complex arrhythmias) were screened, and those who met the conditions signed informed consent before participating. The entire research team consists of clinicians who are familiar with the indications of pacemaker implantation and are proficient in pacemaker implantation surgery and cardiac ultrasound examination. In addition, researchers will receive research-related training. Those willing to participate in the study will be carefully evaluated according to the inclusion and exclusion criteria.
CriteriaInclusion criteria:
a.Meet the 2021 ESC guidelines on cardiac pacing and cardiac resynchronization therapy I or IIa indication for conventional (lead) permanent pacemaker implantation [17, 18]
b.First-time implant of a conventional (lead) pacemaker
c.Preoperative echocardiography showing no, mild, or moderate tricuspid regurgitation
d.Age ≥ 18 years old
e.Able to complete the follow-up study, understand the content of this study, be willing to participate, and sign the informed consent form
Exclusion criteria:
a.Age < 18 years old or > 90 years old
b.Atrial site-paced single-chamber pacemakers (AAI) or patients with implanted defibrillation electrode
c.Have severe tricuspid valve regurgitation, stenosis, or injury before implantation
d.Underwent tricuspid-related procedures prior to implantation
e.A clinical diagnosis of heart failure, transthoracic cardiac ultrasound indicating that the left ventricular ejection fraction is 40% or less, and cannot tolerate pacemaker surgery
f.Other heart disease (including hypertrophic cardiomyopathy, dilated cardiomyopathy, rheumatic heart disease, and endocarditis) or non-cardiac disease that seriously affects the shape or function of the tricuspid valve
g.Acute infectious diseases, important organ injuries, malignant tumors, or have severe diseases and a life expectancy < 1 year
h.Severe liver insufficiency (Child–Pugh level C) and severe renal insufficiency (eGFR < 30 ml/min/1.73 m.2)
i.Abnormal coagulation mechanism and immune function
Informed consentBefore enrollment, professionally trained researchers will introduce the purpose and specific situation of this clinical trial to patients, and patients will also be informed of the possible benefits and potential risks of participating in this trial, to ensure that patients have a full understanding of this trial and have consented to participate in this study at their own discretion. Patients who meet all inclusion criteria without any exclusion criteria will be included after signing written informed consent. All participants’ personal information will always be kept confidential.
Randomization and allocationAccording to the order in which the participants are enrolled, corresponding random numbers will be used for group concealment, and patients will be randomly assigned to the direct group or the bending group. The random allocation table will be held by third-party staff. Before enrollment, the researchers will provide the information of patients who met the enrollment conditions to the third-party staff, and the third-party staff will provide a group number for the patients according to the random allocation table to surgeons (Fig. 2).
Fig. 2
Subject distribution among groups in the study
InterventionsOnce clinical conditions permit, participants will be randomized into two groups: the direct group (“direct-crossing” and “drop-down”) or the bending group (“prolapsing”). In the direct group, the head end of the pacing lead directly crosses the tricuspid valve and enters the right ventricle during the implantation of the pacemaker. In the bending group, the pacing lead is bent during the pacemaker implantation, so that the body of the reflexed lead first crosses the tricuspid valve and then enters the right ventricle. The surgery will be performed by a surgical team who are proficient in pacemaker implantation. Both groups will receive other necessary treatments.
Data handling and record keepingData on all enrolled subjects will be recorded on a case report form (CRF). This task will be the responsibility of the researchers. In addition, subject data will be uploaded to an electronic database for preservation.
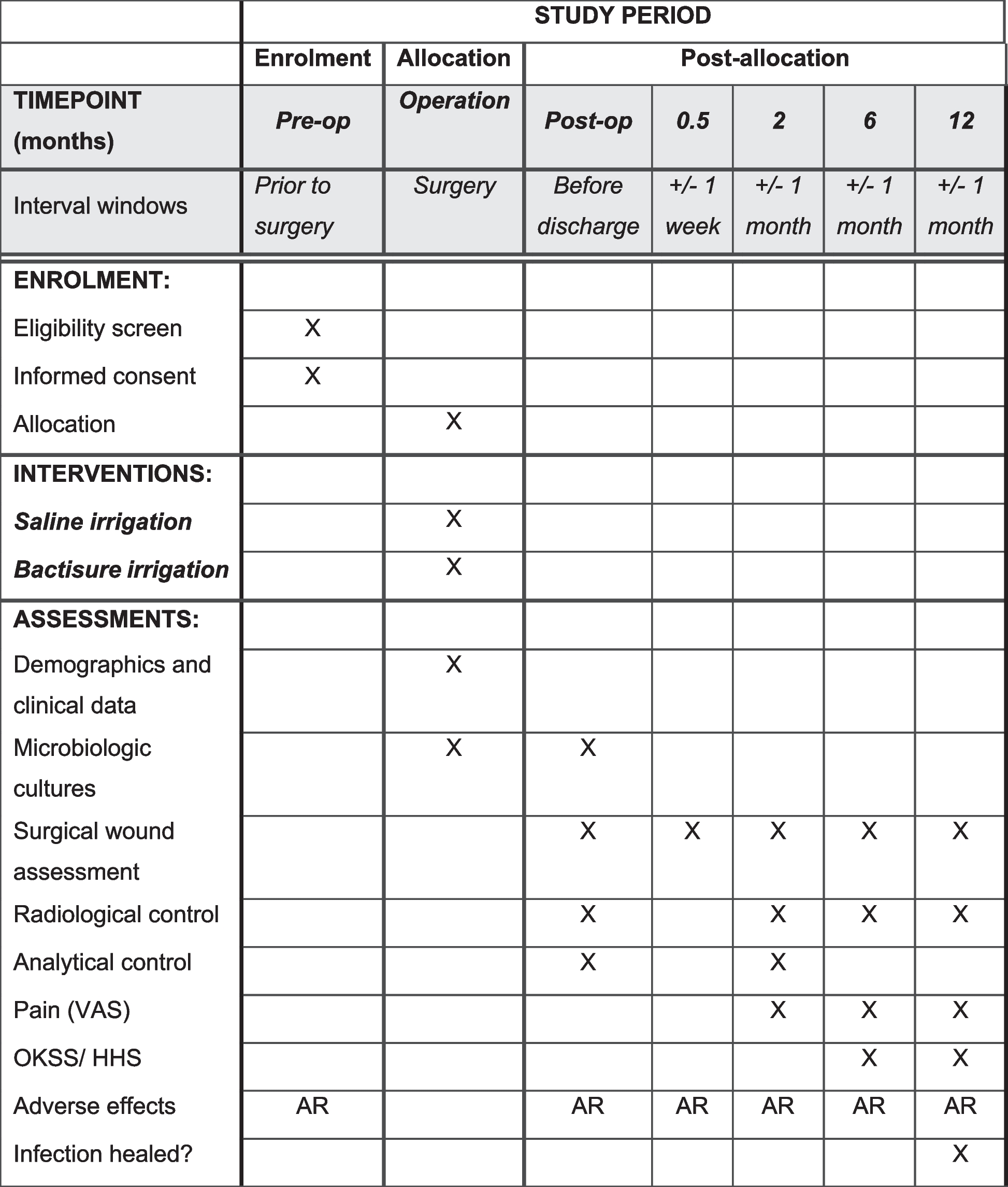
Follow-upFollow-up will be conducted by specific researchers, all of whom will receive training related to follow-up. Enrolled participants will be followed for 1 year. At the 3rd day, 1st, 3rd, 6th, and 12th month after operation, complete echocardiography will be performed to evaluate the degree of tricuspid regurgitation, the positional relationship between the electrode lead and the tricuspid valve complex, and the echocardiography-related indicators including left atrium (LA), right atrium (RA), left ventricle end-systolic diameter (LVs), left ventricle end-diastole diameter (LVd), right ventricle (RV), interventricular septum (IVS), left ventricular posterior wall (LVPW), fractional shortening (FS), and left ventricular ejection fraction (LVEF). In addition, a pacemaker technician will evaluate the function of the pacemaker and adjust the parameters of the pacemaker if necessary to ensure the normal operation of the pacemaker. All-cause mortality, incidence of major cardiovascular and cerebrovascular adverse events (including acute heart failure, malignant arrhythmia, cardiogenic shock, non-fatal myocardial infarction, non-fatal stroke), and whether a second pacemaker implantation operation is required during follow-up will be also recorded by interview or telephone follow-up. The detailed study schedule is listed in Fig. 3.
Fig. 3
Schedule of enrollment, interventions, and assessment for the trial
Plans to promote participant retention and complete follow-upThis study has special follow-up personnel who will notify the patients through telephone, e-mail, or other media in advance of the follow-up time.
WithdrawalPatients may withdraw from the study for any reason. Patients who experience the following conditions will also withdraw from the study:
1.Serious adverse events (AEs) occurred throughout the entire trial process.
2.Unable to continue cooperating with clinical examinations and follow-up due to unexpected reasons.
3.Other situations: if the patients is no longer eligible to participate in the study for reasons at the discretion of the investigator.
Sample sizePreliminary data suggest that electrode-related tricuspid regurgitation occurs in 25% of patients. We predict that the bending group will have reduced incidence of electrode-related tricuspid regurgitation at an incidence of approximately 12.5%, compared to no change in the direct group. In order to obtain 80% power to demonstrate a statistically significant difference between the two transvalvular modalities (two-sided test, α = 0.05), we expect to recruit 300 participants, 150 in the direct group and 150 in the bending group. Given the possibility of exclusion or lost follow-ups, an increase of 20% will be required for the sample size, bringing the sample size to 188 per arm for a total of 376 participants to be enrolled. All calculations are being done with PASS software (NCSS, America, version 11), using the log-rank test, Freedman method.
Data collection and managementAt enrollment, we will collect demographic data from participants, including age, gender, history of disease, cardiac function class (NYHA), NT-proBNP, echocardiography-related indicators including LA, RA, LVs, LVd, RV, IVS, LVPW, FS, and LVEF, and complications after pacemaker surgery (infection, puncture hematoma, pericardial effusion, etc.). To promote data quality, all trial investigators, outcome assessors, and data analysts received relevant training. All data will be repeatedly entered into the database through EpiData software (Denmark, version 3.1) and stored in the scientific research platform of the First Affiliated Hospital of Shantou University Medical College, which is a secure server with access restrictions and supports anonymity, analysis, and review.
Outcome measurementsPrimary outcome: new tricuspid regurgitation or exacerbation of existing tricuspid regurgitation within 1 year after pacemaker surgery.
Secondary outcomes: all-cause mortality during follow-up, incidence of major cardiovascular and cerebrovascular adverse events during follow-up (including acute heart failure, malignant arrhythmia, cardiogenic shock, non-fatal myocardial infarction, non-fatal stroke), and whether a pacemaker implantation operation will be required again during the follow-up period.
Statistical and analytical plansA comprehensive statistical analysis of the entered data will be performed before the database is closed. All data analyses will be performed according to the intent-to-treat (ITT) principle. Among them, categorical variables will be analyzed using the chi-square test, and the results will be expressed as frequencies and percentages. Missing data will be supplemented according to the most frequently occurring value according to the principle of mode in statistics. For continuous variables, the Kolmogorov–Smirnov test will be used to confirm normality. A P > 0.05 will indicate that the data is normally distributed, and the t-test will be used for analysis. If P is less than 0.05, it will mean that the data does not conform to a normal distribution, and advanced raw data transformation (such as logarithmic transformation) will be performed to further check whether the data conform to a normal distribution. If not, the rank sum test will be used for analysis. Results will be presented as mean ± standard deviation. Missing values will be filled with the object’s mean. A P < 0.05 will be used to indicate statistical significance. SPSS statistical software (SPSS Inc., Chicago, IL, USA, version 23) will be used for statistical analysis.
MonitoringThe study will be conducted in accordance with an approved protocol. The Data and Safety Monitoring Committee of the First Affiliated Hospital of Shantou University Medical College will review and interpret the data generated by the study to ensure the safety of the participants and the integrity of the study data. The committee consists of five independent researchers independent of the funders and has no conflicts of interest with this study. Data and safety monitoring committees will review data at 25%, 50%, and 75% post-enrollment to monitor study progress and any possible adverse events (AEs). A computer-generated and time-stamped audit trial will also be implemented to track changes to electronic source documents to ensure the integrity of the research data.
AuditingReview by ethics committee representatives during the research process will be conducted in accordance with national regulations. All protocol modifications will be communicated to interested parties.
Safety and adverse eventsThe main adverse reactions associated with pacemaker surgery include arrhythmia, local hematoma, and infection. Participants who experience any adverse events will receive appropriate treatment from their physicians according to the current clinical practice. Adverse reactions will be checked at each visit. For any adverse reactions that occur, all details, including time of onset, symptoms and signs, extent, duration, laboratory findings, treatment, outcome, and causal relationship to treatment, will be recorded in the CRF. Serious adverse reactions will be reported to the Research Ethics Committee within 24 h, which will decide whether any additional measures should be taken.
AmendmentAll proposed protocol changes will be documented in protocol amendments and will be submitted to ethics committees and regulatory agencies for approval.
留言 (0)