
記住我

Initially, the I CEA study was designed as a multicenter superiority single-blind (independent assessor) randomized clinical trial with 18 months follow-up. After inclusion, patients were randomized 1:1:1 to one of three treatment arms, starting with either (1) NSAID, (2) MTX, or (3) baricitinib.
In arm 1, if patients did not achieve clinical remission (disease activity score (DAS) ≤ 1.6) at 3 months or in case of loss of clinical remission, patients were randomized to arm 2 (MTX) or arm 3 (baricitinib. If they did achieve remission, the NSAID was stopped. In arm 2, if patients did not achieve clinical remission at 3 months or in case of loss of clinical remission on MTX, patients stopped MTX and followed the treatment protocol of arm 3 in which they switched to baricitinib. If they did achieve remission, they continued MTX treatment for 9 additional months. In arm 3, if clinical remission is achieved after 3 months of treatment with baricitinib, patients were randomized to either stop baricitinib immediately or continue baricitinib for 6 more months and then stop baricitinib if the patient is in clinical remission. If after 3 months of baricitinib clinical remission is not achieved or in case of loss of clinical remission while on baricitinib (after 3 months), baricitinib was stopped and patients followed the treatment protocol of arm 2 (switch to MTX). In all treatment arms, if clinical remission was not achieved on MTX nor on baricitinib, subsequent treatment steps aiming at clinical remission were based on shared decision-making between the patient and the treating rheumatologist.
In the original trial design, the trial had two co-primary outcomes: percentage in clinical remission at 3 months and percentage in sustained drug-free remission at study end.
Changes to the study protocolIn October 2022, the European Medicines Agency (EMA) issued a formal safety warning about the use of Janus kinase inhibitors (JAKi) including baricitinib in patients with chronic inflammatory disorders [17]. It was recommended that in patients with an increased risk of major cardiovascular problems or cancer, those > 65 years, and those who smoke or have done so for a long time in the past, JAKi should only be used if no suitable treatment alternatives are available [18]. Based on this warning, it was decided that the use of baricitinib in the I CEA trial as initial treatment in high-risk patients (as defined in the EMA warning) was no longer appropriate. Therefore, it was decided to adjust the exclusion criteria. High-risk patients on baricitinib had to stop treatment with baricitinib and continued with observational follow-up. Additional high-risk patients were no longer included in the trial. This resulted in an estimated exclusion of 2/3 of the target patient population and a considerable delay in patient inclusion. At this point, we also performed a blinded interim analysis (see section on Sample size analyses) to assess outcomes at 3 months. Based on the blinded interim analysis, changes in the exclusion criteria, and the slow inclusion rate, we adjusted the primary outcome measures and simplified the trial design.
The primary outcome measure was adjusted to the change in disease activity at 3 months (also see the section on Sample size calculation), the follow-up duration was limited to 12 months and the time on study medication was limited to the first 3 months (for a more extensive description see the Current design section).
Eventually, the slow inclusion rate led to the decision to stop further patient inclusion after 111 patients had been included, but to finish follow-up for the patients already in the trial. This decision was made by the study sponsor in consultation with the independent medical doctor and medical ethics committee, independently of the funder. This decision was supported by an updated sample size calculation (see section on Sample size calculations) which takes into account adjustment for baseline DAS, showing that we would still be able to demonstrate a difference between the treatment arms that are expected to differ most in treatment effects. These modifications to the protocol have subsequently been approved by the medical ethics committee and communicated with the data safety monitoring board, participating hospitals, patient research partners, and trial participants. The updated protocol has been finalized prior to unblinding data for the primary analysis.
Current trial statusBecause of the recent change to the trial protocol and the earlier than expected ending of patient inclusion, it was not possible to publish the protocol before patient inclusion ended. Follow-up of patients is ongoing and planned until the end of 2024.
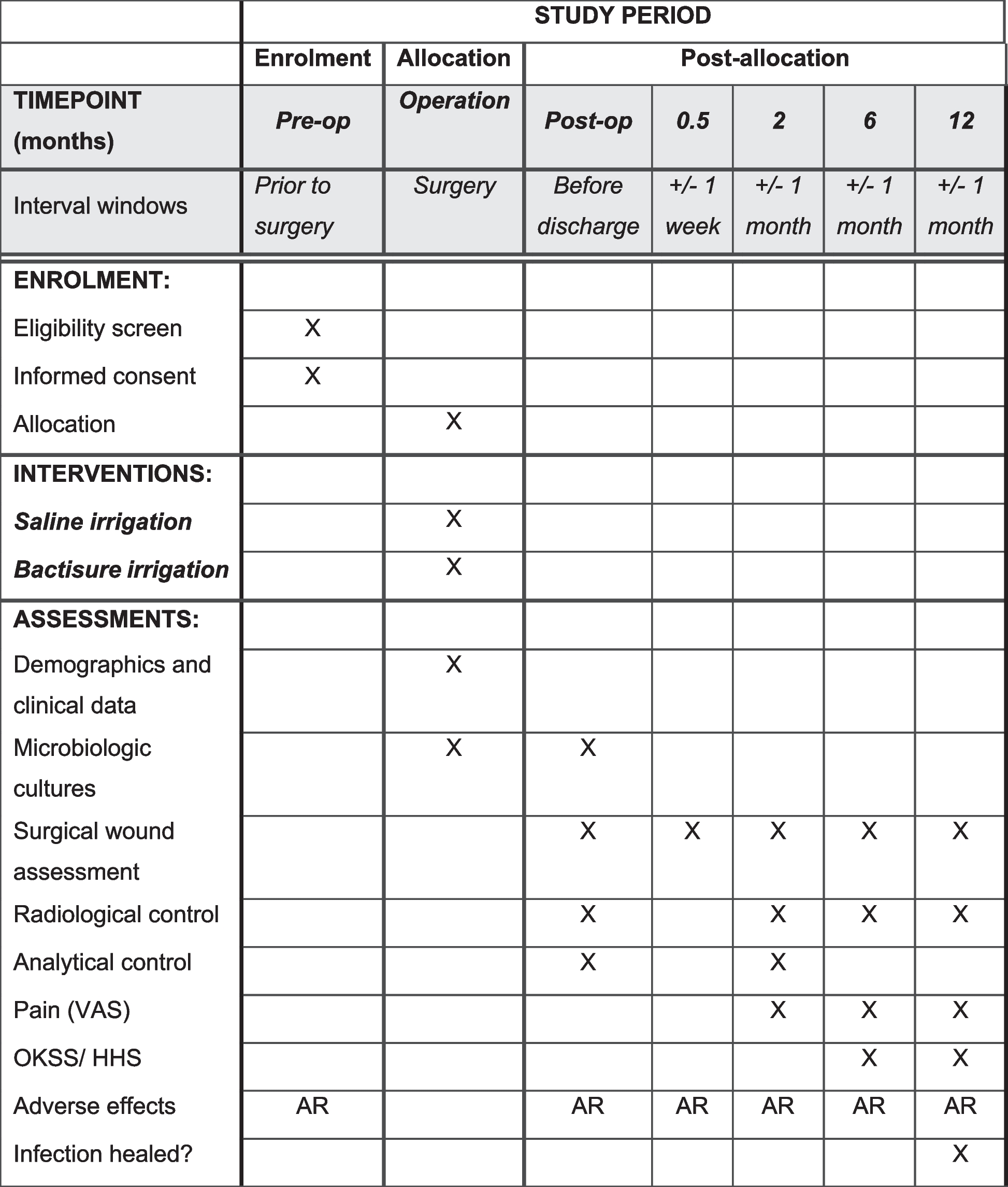
Current design of the Induction of Cure in Early Arthritis (I CEA) trialThe I CEA trial is a multicenter superiority investigator-initiated single-blind (independent assessor) randomized clinical trial with a 3-month interventional and 9-month observational follow-up period in patients with early (< 1 year symptom duration) DMARD naïve undifferentiated arthritis (Fig. 2). After inclusion, patients will be randomized 1:1:1 to one of three treatment strategies:
1)Symptom relief with analgesics, NSAIDs, and an intramuscular or intra-articular injection of glucocorticoids, and wait for spontaneous remission;
2)Start with MTX and a single intramuscular or intra-articular injection of glucocorticoids; analgesics and NSAIDs optional;
3)Start with baricitinib and a single intramuscular or intra-articular injection of glucocorticoids; analgesics and NSAIDs optional;
Fig. 2
SPIRIT figure. Abbreviations: NSAID, non-steroidal anti-inflammatory drug; RR, respiration rate; P, pulse; temp, temperature; TBC, tuberculosis; MTX, methotrexate; SHS, Sharp/van der Heijde score; HAQ, health assessment questionnaire; SF-36, short form 36; MACTAR, McMaster-Toronto Arthritis Patient Preference Disability Index; EQ5D-3L, EuroQol-5 dimension-3 levels; HADS, hospital anxiety and depression scale; IPQ-K, Illness Perception Questionnaire-short; VAS, visual analog scale [1]. Hb, total WBC, absolute neutrophil count, absolute lymphocyte count, thrombocyte count, serum creatinine, AST, ALT, total cholesterol, HDL, and LDL [2]. erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) [3]. Anti-citrullinated protein antibodies (ACPA, including isotypes), Rheumatoid Factor, anti-carbamylated protein antibodies (anti-CarP), and other potentially newly discovered predictors of outcome [4]. TBC screening will be performed according to local guidelines in the participating center. In the LUMC, in which most patients have been included, this includes the Mantoux test (PPD) and Quantiferon [5]. X-rays are required if they were not performed and evaluated within approximately 6 months prior to screening [6]. joints presenting with clinical arthritis will be X-rayed for potential damage (progression) at baseline and, independent of the persistence of local clinical arthritis, at 12 months. In case of progression of arthritis, joints that become newly inflamed during the study may also be X-rayed if clinically relevant, but need no systematic follow-up X-ray at 12 months. * In exceptional cases study visits at weeks 2, 4, and 8 can be carried out by phone at which the research nurse will ask for pain, swelling, flexion, or extension limitation
The study will be performed in 2 academic and 5 peripheral hospitals in the Netherlands (Supplementary file 1 l).
Eligibility criteriaInclusion criteria≥ 18 and < 65 years of age and able to give written informed consent (in Dutch or English) and fill out questionnaires in Dutch (or English version, if available)
Clinical early unclassified arthritis of at least 2 joints, not fulfilling ACR/EULAR 2010 criteria for RA (score < 6, see Table 1).
Symptom duration of arthritis < 1 year
No other plausible cause for arthritis (including infection, (pseudo-)gout, psoriatic arthritis, non-rheumatoid auto-immune disease, paraneoplastic arthritis)
Disease Activity Score (DAS) > 1.6 and at least two swollen and painful joints
DMARD naïve (prior anti-malarials discontinued more than 4 weeks ago are allowed)
No wish to become pregnant, breastfeed or father a child during the study
No contraindications to treatment with NSAIDs, MTX (or sulfasalazine or leflunomide as an alternative), and baricitinib as required in the study.
Table 1 2010 ACR/EULAR classification criteria for RA [12]Exclusion criteriaAlcohol or substance abuse
Current smoking or a long history (≥ 20 years, stopped < 10 years ago) of smoking
Immuno-compromised state either based on co-morbidity or co-medication
Leucopenia < 3 × 109/l, and/or neutropenia < 1 × 109/l
Hemoglobin < 5 mmol/l
Increased liver enzymes > 3 × upper limit of normal
Renal insufficiency with estimated creatinine clearance < 60%
Interstitial lung disease as seen on chest radiograph
Maintenance treatment with glucocorticoids exceeding prednisone 10 mg daily or equivalent (inhalation corticosteroids excepted).
Active or ongoing chronic infection, (recurrent) serious infection(s) in past 4 months, latent TB with refusal of anti-tuberculous treatment, hepatitis B with positive DNA viral load or hepatitis C with positive RNA viral load, patients with anti-HB2 and anti-HBc antibodies with refusal to monitor hepatitis B DNA expression
Increased risk for arterial or venous thrombosis or increased risk for major cardiovascular events, as assessed by the treating rheumatologist. Risk of arterial thrombosis is based on the calculation of the “cardiovascular SCORE,” which includes cholesterol ratio, systolic blood pressure, age, gender, diabetes, history of cardiovascular disease, and smoking status. Only patients in the “green” category of the risk table will be included [19].
Increased risk of malignancy; this includes patients with a history of malignancy (except basal cell carcinoma, squamous cell carcinoma, cervical carcinoma in situ, and other malignancies that have been treated curatively > 10 years ago, for instance, types of thyroid cancer) and patients with a syndrome which increase cancer risk (e.g., BRCA/HNPCC/LYNCH).
The exclusion of patients who fulfill the 2010 classification criteria for RA implies that the study population will be mostly seronegative, although potentially with many active joints and significant symptoms and limitations. Also, seropositive patients with very few inflamed (large) joints, normal CRP and ESR, and/or symptom duration < 6 weeks will qualify for participation in the study.
InterventionPatients will be randomized to one of three types of treatment, each combined with a single parenteral dose of glucocorticoids. Each treatment will be given during a period of 3 months. After this period, patients will enter an observational period of 9 months in which they will be treated according to the shared decision of the rheumatologist and patient. During this period, the treating physician is allowed to discern the most appropriate treatment from all available treatment options, including DMARDs.
Arm 1: start or continue an NSAID (or COXIB) in a usual daily dose. The choice of NSAID or COXIB is left to the shared decision between the rheumatologist and patient. This can be for example naproxen 2 dd 500 mg or equivalent such as ibuprofen 2 dd 600 mg (maximum 2 dd 800 mg) or diclofenac 2 dd 75 mg, or celecoxib 2 dd 100 mg. The use of paracetamol with a maximum dose of 3 dd 1000 mg is optional. On indication drugs for gastro-protection will be added. Patients will receive a single dose of intramuscular or intra-articular glucocorticoids at the start of treatment (40 mg methylprednisolone, 40 mg triamcinolone, or alternative glucocorticoid in an equivalent dose). In the case of intra-articular injections, these are not restricted to a maximum number of injectable joints, as long as the total dose does not exceed 40 mg triamcinolone. Smaller joints may receive lower doses of triamcinolone (or equivalent).
Arm 2: start with MTX 15 mg/week increased to 25 mg/week in 4 weeks or the maximum tolerated dose, orally or subcutaneously. If MTX is contraindicated, sulfasalazine (starting dose 500 mg/day, increased in 4 weeks to 1000 mg twice daily) or leflunomide (20 mg/day or 10 mg/day depending on tolerance) can be provided as alternatives. This is in accordance with the EULAR recommendations for the management of RA [5]. Patients will additionally receive a single dose of intramuscular or local intra-articular glucocorticoids (options as in arm 1) at the start of treatment.
Arm 3: start with baricitinib 4 mg/day. Patients will receive a single dose of intramuscular or local intra-articular glucocorticoids (options as in arm 1) at the start of treatment.
In arms 2 and 3, co-treatment with an NSAID or COXIB with or without paracetamol (options as in arm 1) is allowed but not required. In all arms, in case of a disease flare a single additional intra-articular or intra-muscular injection of glucocorticoids (dose as in arm 1) is allowed in case of insufficient improvement on the initial (randomized) treatment, provided the next study visit is > 1 month in the future.
Oral glucocorticoids are only allowed during the trial if they are prescribed as part of stable maintenance treatment for an underlying condition (e.g., adrenal insufficiency) and the dosage does not exceed 10 mg/day.
Baricitinib is registered for use in patients with RA who had an inadequate response to MTX or another DMARD. The approved dose in the EU for the treatment of RA is 4 mg/day orally, which will also be used in the current study [20]. Baricitinib is currently not approved or registered for the treatment of UA patients. If during the treatment period of the study, patients develop manifest risk factors for continued use of baricitinib as described by EMA [17], baricitinib may no longer be prescribed, and an alternative treatment will be chosen based on shared decision-making between the patient and treating rheumatologist. Also, baricitinib treatment will be interrupted during periods of immobilization or surgery to reduce the risk of thrombosis.
Informed consentInformed consent will be obtained by local rheumatologists, research nurses or study doctors, who are Good Clinical Practice certified. Written informed consent will be obtained from all eligible patients before they are randomized. As part of the informed consent procedure, patients are asked whether they provide permission to:
◦ Request their cause of death from the Dutch Central Bureau of Statistics in case they die during the study
◦ Store their data and body materials after the trial to use for future studies
◦ Perform DNA research related to the cause of joint inflammation or adverse events.
◦ Be approached for future studies
◦ Store their name and address (separately from the study data) to receive newsletters and other communications.
A model consent form can be found in Supplementary file 2.
OutcomesPrimary outcomesDAS is a validated composite measure representing response to therapy in R and UA patients. DAS is based on a 44 swollen joint count including a 53 joint Ritchie Articular Index [21].
Calculation of DAS: DAS-4(ESR) = 0.54*√(RAI) + 0.065*SJC44 + 0.33*ln(ESR) + 0.007*GH.
Secondary outcomes◦ Percentage in clinical remission at 3, 6, 9, and 12 months
◦ Time to clinical improvement, through patient diary (pain, stiffness, fatigue).
◦ Percentage loss of clinical remission (DAS < 1.6) at 6, 9, and 12 months
◦ Time to flare, defined as loss of remission
◦ Functional ability over time as measured by HAQ-DI [22].
◦ Disease activity over time as measured by DAS
◦ Progression to classifiable (ACR/EULAR 2010) RA over time [5].
◦ Toxicity, defined by the number and severity of adverse events.
◦ Pain, measured by the Pain Detect Questionnaire (PD-Q) and a VAS scale from 0 to 10 cm
◦ Disease activity as assessed by treating rheumatologist on a VAS scale from 0 to 10 cm
◦ Morning stiffness, measured by a VAS scale from 0 to 10 cm
◦ Fatigue, measured by a VAS scale from 0 to 10 cm
◦ Physical and emotional wellbeing, measured by the Short Form 36 [23].
◦ Quality of life as measured by the 3-level EuroQol-5 dimensions (EQ-5D) [24].
◦ (Progression of) radiologic damage (Sharp/van der Heijde score) at 6 and 12 months.
Exploratory outcomes◦ Illness perceptions, measured by the Brief Illness Perception (IPQ-K) questionnaire
◦ Cost-utilities, based on healthcare use, costs, and work productivity assessed in patient diaries and the EQ-5D questionnaire.
◦ Patient satisfaction with treatment, measured by visual analog scales (VAS) from 0 to 10 mm
◦ Depressive feelings, measured by the Hospital Anxiety and Depression Scale (HADS)
◦ Daily functioning in preferred activities, measured by the McMaster-Toronto Arthritis Patient Preference Disability—Questionnaire (MACTAR) [25, 26].
Remission definitionDefinition of clinical remission depends on a decision tree:
DAS by independent assessor < 1.6: defined as clinical remission.
DAS by independent assessor ≥ 1.6: assessment by treating rheumatologist required.
If the rheumatologist finds clinical arthritis: defined as no clinical remission.
If not: defined as clinical remission.
This extra evaluation was introduced to prevent treatment escalation for high DAS based on pain without clinical arthritis.
At all visits, both the blinded assessor and the treating physician perform an independent joint examination. The treating rheumatologist is allowed to disregard the joint assessment of the blinded assessor in case of disagreement. The reason for disagreement and treatment decisions are recorded.
Study diary and drug accountabilityParticipating patients will be asked to fill in a study diary where they will record medication use, as well as symptoms and costs.
The diary includes:
◦ Daily numeric rating scales (0–10 mm) for pain, fatigue, and stiffness.
◦ Work Productivity and Activity Impairment Questionnaire (WPAI)
◦ Healthcare use, including visits to rheumatologists, general practitioners, physical therapists, or others, related to arthritis or side effects of study medication
◦ Additional expenses due to joint complaints, including alternative healthcare, travel costs, assistive devices (e.g., brace, splint), domestic help, use of over-the-counter or prescribed medication such as analgesics, etc.
◦ Adverse events: type and frequency.
The diaries are reviewed by the research nurse at each study visit. Diaries are completed digitally and recorded in the Case Record Form. At each study visit, patients will be required to bring the study medication for drug accountability. The research nurse or pharmacy will register any remaining study medication in the Case Record Form and take in and return any unused medication to the trial pharmacy at the end of study participation.
Data collection forms can be obtained upon request from the corresponding author.
Adverse event reportingAdverse events (AEs) are defined as any undesirable experience occurring to a subject during the study, whether or not considered related to the investigational product/trial procedure/the experimental intervention. All AEs reported spontaneously by the subject, observed by the investigator or his staff, or detected during routine laboratory testing as required for study medication, will be recorded.
A serious adverse event (SAE) is any untoward medical occurrence or effect that:
◦ Results in death;
◦ Is life-threatening (at the time of the event);
◦ Requires hospitalization or prolongation of existing inpatients’ hospitalization;
◦ Results in persistent or significant disability or incapacity;
◦ Is a congenital anomaly or birth defect; or
◦ Any other important medical event that did not result in any of the outcomes listed above due to medical or surgical intervention but could have been based upon appropriate judgment by the investigator.
An elective hospital admission will not be considered as an SAE, unless it was prolonged or considered to be related to the study drug.
All participating centers will report SAEs to the organizing center at the LUMC and provide additional information as requested. The organizing center will report to the relevant authorities. In case of SAEs or SUSARs related to the use of baricitinib, the pharmaceutical company will also be notified.
All AEs will be followed until they have abated, or until a stable situation has been reached. Depending on the event, follow-up may require additional tests or medical procedures as indicated, and/or referral to the general physician or a medical specialist. SAEs will be reported till the end of the study.
ProceduresThe SPIRIT table in Fig. 2 describes the time schedule of enrollment, clinic visits and intervention assessments.
Study visits will be every 3 months, with additional visits at weeks 2, 4, and 8 for clinical assessment only (no treatment adjustments). At each 3-monthly study visit, the DAS will be calculated based on a full joint assessment by the independent assessor and ESR/CRP as measured in a 7.5-ml blood sample that has been collected at baseline and thereafter within 1 week before the three-monthly study visits. An additional 3.2 ml (4 × 800 µL) sample will be stored and used for retrospective investigations into potential predictors of outcome, including non-routine auto-antibodies, cytokines (at screening, 3 months, and at end of study (12 months or earlier in case of a premature stop) and 1 EDTA sample will be taken at screening to identify DNA profiles potentially associated with disease or treatment outcomes. Plain radiographs of hands (per hand, one direction) and feet (per 2 feet, two orientations) will be made at screening and at 6 and 12 months, as well as of other joints with arthritis at baseline and at the end of the study (12 months or earlier). At each three-monthly study visit, questionnaires will be presented for filling in either online via a secure webpage or as a paper version, later to be uploaded in the database.
Based on the three-monthly DAS and the absence or presence of clinical arthritis, the treating rheumatologist will need to adjust the medication as required in the study protocol. The rheumatologist will fill out a small questionnaire on, yes/no agreement with DAS, and yes/no agreement with the next treatment step and preferred alternative treatment if outside a study protocol.
Safety monitoringRequired safety monitoring prior to treatment initiation with baricitinibBefore therapy initiation, patients should be screened for latent tuberculosis and viral hepatitis. In this study, latent tuberculosis and/or active hepatitis rules out participation. Tuberculosis screening will be performed according to local guidelines in the participating centers. In the unusual case of a patient with anti-HBs and anti-HBc but without HBsAg, the patient may participate but should stop baricitinib and will be very closely monitored for expression of hepatitis B-DNA in collaboration with a hepatologist.
Baseline laboratory monitoring should be performed to determine absolute lymphocyte count (ALC), absolute neutrophil count (ANC), and hemoglobin values. Baricitinib should be avoided in patients with active serious infection and is not recommended in patients with a total leucocyte count less than 0.5 × 109/l, neutrophil count of less than 1 × 109/l, hemoglobin less than 5 mmol/l, glomerular filtration rate of < 60 mL/min/1.73 m2, or severe hepatic impairment. There may be an increased risk for thromboembolic events, particularly in patients with a history of thromboembolic events.
Should patients develop lymphopenia, neutropenia, or anemia during treatment, depending on severity discontinuation or dose adjustments may be considered.
Required safety monitoring during treatment with baricitinibEvery 3 months, complete blood count with differential, renal and liver function tests (creatinine, AST, and ALT), lipid panel (total cholesterol, HDL, and LDL), and infection monitoring (ESR and CRP) are recommended. Those at increased risk for skin cancer should receive routine skin examinations.
Safety monitoring before and after the start of methotrexateBefore the start of methotrexate: chest X-ray total blood count, liver enzymes (AST and ALT), renal function (creatinine and estimated creatinine clearance).
After the start of methotrexate: every 3–4 weeks total blood count, liver enzymes (AST and ALT), renal function (creatinine and estimated creatinine clearance).
With continued use of methotrexate: every 3 months total blood count, liver enzymes (AST and ALT), renal function (creatinine and estimated creatinine clearance).
Data safety monitoring board (DSMB)A DSMB committee including a clinical statistician and two rheumatologists who have no conflict of interest regarding the study or the investigational product, will meet every 6 months. The DSMB will review toxicity data and will advise the sponsor on the continuation of the study and potential adjustments to the protocol in case of safety issues.
The advice(s) of the DSMB will only be sent to the PI of the study. Should the PI decide not to fully implement the advice of the DSMB, the sponsor will send the advice to the reviewing medical ethics committee, including a note to substantiate why (part of) the advice of the DSMB will not be followed.
Data management and confidentialityData and documents will be collected at the coordinating center and stored securely under pseudonymized patient code to maintain participant confidentiality and conform to GCP guidelines. As much as possible, and as far as privacy requirements allow, data will be collected digitally only using the validated data capture software CASTOR [27], either from home by the patient or during the study visits by the research nurse. Digital data will be stored in a designated folder on the local network, which is only accessible by authorized processors. A local backup is regularly made. Additional data will be collected at each study visit by the research nurse and stored at the coordinating center. Any non-digital data will be stored securely at the study site. All records that contain names or personal identifiers, such as informed consent forms, will be stored separately from study records identified by patient codes. These will be stored in a separate, locked file in an area with limited access. Blood samples for non-routine tests will be drawn and stored at the local center. Samples will be stored under pseudonymized patient code for up to 25 years after study completion or until the patient requires them to be destroyed. The use of samples is limited to investigations as mentioned in this protocol. Only the study database manager will have access to the code in relation to personal data. LUMC monitors and the Health and Youth Care Inspectorate (IGJ) have access to the source data for data verifying purposes. All members of the study team are certified and regularly trained regarding GCP and GDPR guidelines and national and international laws and regulations.
Participants’ study information will not be released outside of the study without the written permission of the participant.
MonitoringAllocated study personnel based at the coordinating center will visit the participating centers at three-monthly intervals to monitor study procedures and collect stored blood samples for storage at the coordinating center. Through queries to rheumatologists and/or study nurses, they ensure that the study protocol is followed. In case of protocol deviations, these are registered, including an explanation on why the deviation occurred and with which consequences, correcting if possible. Independent LUMC monitors will visit the participating hospitals to ensure adequate trial procedures.
Allocation and blindingPatients will be allocated 1:1:1 to one of three treatment strategy arms by variable block randomization stratified per center. The randomization and allocation sequence was generated by an external staff member using the computer program Castor [27]. Randomization and enrollment were performed by study doctors, research nurses, or local study coordinators.
The independent assessors but not patients nor treating rheumatologists will remain blind for the allocated treatment arm. Randomization codes will only be broken at the analysis of results (to allow a gatekeeping procedure) regarding the primary study objective, or earlier if deemed inevitable following advice of the Data Safety Monitoring Board and discussion with the central Medical Ethics Committee.
Baricitinib will be provided as study medication by Eli Lilly and stored at the Leiden University Medical Center Trial Pharmacy. Research nurses (blinded assessors) who are involved in the screening and study assessments are blinded for treatment and are, therefore, not allowed to pick up study medication at the pharmacy. They will assess the disease activity and communicate the result to the rheumatologist. Based on the required treatment step, the rheumatologist will issue a prescription for baricitinib as study medication to the trial pharmacy, unblinded personnel will collect the baricitinib, and the rheumatologist will hand it to the patient. Parenteral glucocorticoid injections are standard treatments, available in rheumatology outpatient clinics and will be given by the rheumatologist or a trained nurse. Other medications in the study (MTX or alternative DMARDs, naproxen or alternative NSAIDs or COXibs, paracetamol if necessary) will be as usual prescribed by the rheumatologist and picked up by the patients at the local pharmacy after the study visit.
RetentionTo improve participation and retention, in exceptional cases study visits at weeks 2, 4, and 8 can be carried out by phone at which the research nurse will ask for pain, swelling, flexion, or extension limitation. All patients who withdraw informed consent before the study end will be asked to complete an end-of-study visit and permission to reconnect in the future for additional consent for potential long-term follow-up studies.
Sample size calculationInitial sample size calculationThe initial sample size was calculated based on pairwise comparisons between 3 groups and 2 primary outcomes (% clinical remission at 3 months and % drug-free remission at 18 months) and resulted in a required number of 103 patients per treatment arm.
During the trial, due to new safety guidelines regarding JAK inhibitors—including baricitinib—by the European Medicines Agency, we decided to adjust the exclusion criteria to avoid the inclusion of patients who could not be treated with baricitinib. As a result, approximately 2/3 of the patients with UA were no longer eligible for participation. We performed blinded interim analyses of outcomes so far at month 3.
Blinded interim analysesAt the time of the blinded analyses, 23, 20, and 19 patients in the 3 treatment groups had completed month 3. Remission rates were (random order as groups were unidentified) 35%, 52%, and 53%, whereas 30%, 52%, and 74% were initially hypothesized for planned groups 1–3, respectively. The mean DAS at month 3 in the blind interim analysis was 1.7, 1.8, and 2.3 (random order).
Based on these interim effect sizes, we calculated the sample size required to prove a statistically significant difference in remission rates (p < 0.05). With remission rates observed at month 3 in the interim analysis between 35 and 52.6%, this resulted in a sample size between 146 per treatment arm (best case scenario) and 295,479 per treatment arm (worst case scenario). Therefore, to prevent premature termination of the trial, remission at 3 months and drug-free remission at 18 months as primary outcomes were dropped, and clinical improvement measured as change in disease activity (DAS) at 3 months was chosen as the new primary outcome. In addition, patients with a monoarthritis at baseline were excluded, since these patients often have a very low DAS already at baseline and are therefore less likely to show a significant improvement in DAS at 3 months.
Interim sample size calculationsAt the interim analyses, the observed mean DAS from the two groups that differed most were 2.26 and 1.67. In accordance with the new primary outcome measure ‘change in DAS from baseline to 3 months’, a new sample size analysis based on ANCOVA (taking into account adjustment for baseline DAS) was performed. With an SD of 0.946, (largest SD, anonymized subgroup A) a power of 90%, alpha level of 0.05, and an estimated correlation between baseline DAS and DAS at 3 months of 0.8, this would require 20 patients per group. Repeating this analysis with a mean DAS of 2.26 and 1.78 (the second largest difference in DAS in the interim analysis), this resulted in 30 patients per group.
Since we do not know which of these differences will correspond to the first step in the gatekeeping analysis (Fig. 3), the sample size analysis calculation was based on the second largest difference in DAS, to increase the chance that we will be able to compare multiple treatment groups.
Fig. 3
Gatekeeping analysis: stepwise statistical procedure. Legend: DAS, disease activity score; NSAID, non-steroidal anti-inflammatory drugs
To allow a per-protocol analysis as well as a intention-to-treat analysis, we will include an extra number of patients equal to the number of patients already included who had monoarthritis at the time of inclusion, and to compensate for early drop-out. In the current study population, we expected, and also noticed in the interim analysis, a drop-out rate of 10%. Thus, we anticipate requiring a total of 109 patients (30 × 3 × 1.1 (drop-out rate) + 10 (patients with monoarthritis already included). The sample size analysis was performed with Stata (StataCorp LLC, release 16, command sampsi).
The sample size was determined for the primary endpoint, and sample size calculations do not take into account the number of secondary outcomes that will be assessed. Results from secondary endpoint analyses will be interpreted with caution and in relation to clinical relevance.
Statistical analysesMissing dataMissing data will be handled in the same way in each of the analyses. If less than 5% of the data is missing, we will perform a complete case analysis. If more than 5% of the data is missing, we will perform multiple imputation [28]. Multiple imputation will be performed using chained equations, with predictive mean matching (with 5 observations to draw from) to impute missing data in continuous variables and (multinomial) logistic regression to impute missing data in categorical variables. The number of imputed datasets will be at least equal to the included variable with the largest proportion of missing data. Thus: if one of the included variables has 20% missing data, at least 20 imputed datasets will be created. Variables used for imputation will include primary and secondary outcome measures, baseline characteristics, (components of) disease activity measures or physical functioning that are available over time, treatment arm within the trial, time in follow-up, and hospital of inclusion. Analyses based on variables with missing data will be performed within the multiply imputed database and pooled using Rubin’s rule.
Primary outcomeThe primary objective is to identify the best strategy to ensure the most decrease in mean DAS at month 3. Patients who were included with DAS < 1.6 (remission) will be disregarded from the main analysis. Change in DAS will be operationalized as mean DAS at 3 months adjusted for baseline DAS.
Patients will be analyzed within their original treatment arm (intention-to-treat). Visual analysis of box plots will be used to assess whether outcomes are similar between the different hospitals that included patients. If there are indications for heterogeneity in outcomes between the hospitals, the 3 treatment arms will be compared using Generalized Estimating Equations (GEE) analysis, with DAS at 3 months as outcome, treatment arm as predictor, and hospital of treatment (LUMC vs non-LUMC) as a cluster variable. Should cluster sizes be very heterogeneous or clusters be small in general, additional sensitivity analyses based on re-sampling will be considered (bootstrap, permutations). We will adjust for baseline DAS. If there are no indications for heterogeneity of outcomes across hospitals that include patients, we will perform an ANCOVA analysis with DAS at 3 months as an dependent variable, treatment arm as an independent variable, and baseline DAS as covariate. We will additionally assess whether symptom duration and ACPA status at baseline are prognostic for the outcome and further improve the fit of the models. We will employ a gatekeeping analysis (Fig. 3). We will first compare the baricitinib treatment arm with the NSAID treatment arm. This comparison will be done first because from a clinical point of view, this comparison is expected to have the largest effect. If this comparison is statistically significant (p < 0.05), we will continue with the comparison between the MTX treatment arm and the NSAID treatment arm. If this analysis is statistically significant, we will continue with the final comparison between the MTX treatment arm and the baricitinib treatment arm.
For the primary outcome measure we will conclude that early treatment (with MTX or baricitinib) is not proven to have an advantage over delayed treatment if both the MTX and the baricitinib arms are (in separate steps of the gatekeeping procedure) not statistically significantly different from the NSAID treatment arm). If both the MTX and baricitinib arms perform statistically significantly better than the NSAID treatment arm, without a statistically significant difference between the MTX and the baricitinib arm, we will conclude that early treatment with either of the drugs is better than delayed treatment and a preference for one of these drugs may follow from the secondary outcomes (e.g., cost-utility and adverse events). If we find that either the MTX or baricitinib arm performs statistically significantly better than the other arm (and better than NSAID treatment), we conclude that there is a preference for treating early with this drug (MTX or baricitinib) in terms of efficacy.
Secondary outcomesFor the secondary outcome measures p < 0.05 will be considered statistically significant.
Observed radiographic progression based on the Sharp/van der Heijde score at 6 and 12 months compared to baseline will be graphically depicted in a cumulative probability plot for each treatment arm. Both the number of patients with (S)AEs and the number of (S)AEs will be presented by the treatment arm. The number of AEs and SAEs will be calculated per 100 patient years. AEs and SAEs, Hospital Anxiety and Depression scale, and the HAQ, and other continuous outcomes will be compared between treatment arms at 3 and at 12 months. Radiographic progression and the proportion of patients that progressed to classifiable (2010 criteria) RA will be compared between treatment groups at 12 months. The HAQ and the radiographic progression scores will be additionally assessed over time. Also, other continuous variables, including the SF36, the VAS scores for local joint pain, general fatigue, general functional disability, and physician assessment of disease activity will be assessed over time and by treatment arm.
For the comparison of the 3 treatment arms differences between variables at fixed timepoints will be compared using GEE clustered by the hospital (LUMC vs. other hospitals) or by ANCOVA (with similar reasoning as for the primary outcome) with NSAIDs as reference arm. In case both MTX and baricitinib are statistically significantly better than NSAIDs, the analysis will be repeated with MTX as a reference arm.
The comparison of treatment groups over time will be performed using GEE, taking into account repeated measurements within individuals over time. To define how time will be treated (e.g., continuous or categorical) visual inspection of graphs depicting each outcome over time for the different treatment arms will be performed.
Time to progression to classifiable RA (according to 2010 criteria), time to clinical remission, and time to flare for patients achieving remission for the first time will be compared using a Kaplan–Meier curve and a log-rank test. The relationship between the treatment arm and all described time-dependent variables will be analyzed with Cox regression, potentially corrected for ACPA, DAS, and symptom duration.
Sensitivity analyse
留言 (0)