Plasmid vaccines
The HTNV DNA vaccine plasmid, pWRG/HTN-M (co), was constructed by cloning cDNA representing the HTNV M segment open reading frame (ORF) which encodes Gn and Gc, optimized for human codon usage, into the NotI and BglII-restriction sites of pWRG7077 as described previously31. The PUUV DNA vaccine plasmid, pWRG/PUU-M(s2), was constructed similarly using cDNA that was engineered as a consensus sequence of several PUUV isolates, and optimized for human codon usage and mRNA stability (GeneArt, Regensburg, Germany)9. The HTNV and PUUV DNA vaccines were produced under current good manufacturing practices (cGMP) by Althea Technologies, Inc. (San Diego, CA, USA). The vaccine was formulated at 2 mg/mL in phosphate-buffered saline (PBS) (Thermo Fisher Scientific, Waltham, MA, USA). The potency of the DNA vaccines was measured by evaluating the expression of the hantavirus glycoproteins using a standardized flow-cytometry-based in vitro potency assay performed essentially as described previously32. For the combined HTNV/PUUV vaccine, on the day of vaccination, a simple 1:1 mixture of both vaccines was prepared by combining equal volumes of HTNV and PUUV DNA vaccines in a separate vial.
PharmaJet system
The clinical use of the PharmaJet Stratis NFIS (PharmaJet Golden, CO, USA) has been previously described, including for an ANDV DNA vaccine Phase 125. Stratis is an FDA 510k-cleared device that delivers a 0.5 mL jet of liquid at high pressure that penetrates the skin into the muscle.
Clinical study subjects
Healthy adult volunteers, male and female, between the ages of 18 and 49 (inclusive) were recruited through the Walter Reed Army Institute of Research (WRAIR) Clinical Trials Center, Silver Spring, Maryland. All recruiting and consent methods and materials were compliant with current good clinical practice (GCP) guidelines and approved by the Walter Reed Army Institute of Research (WRAIR) Institutional Review Board (IRB). All study procedures took place at this site.
Data obtained from all subjects who received at least one vaccination were included in the safety statistical analysis. Exclusion criteria included pregnant or lactating females, a history of severe reactions to any vaccination or a history of severe allergic reactions, acute or chronic medical or psychiatric conditions, receipt of another vaccine or IND product within 30 days of the planned first dose, receipt of blood products within 120 days prior to enrollment, immunosuppressive or immunodeficient conditions and chronic use of immunosuppressive drugs other than inhaled or topical steroids. To exclude persons with possible prior exposure to a hantavirus, serum samples from all subjects were screened at a 1:20 dilution for pre-existing antibodies to HTNV and PUUV using HTNV and PUUV pseudovirion neutralization assays (PsVNAs). Only seronegative subjects were enrolled in the study.
Clinical study design overview
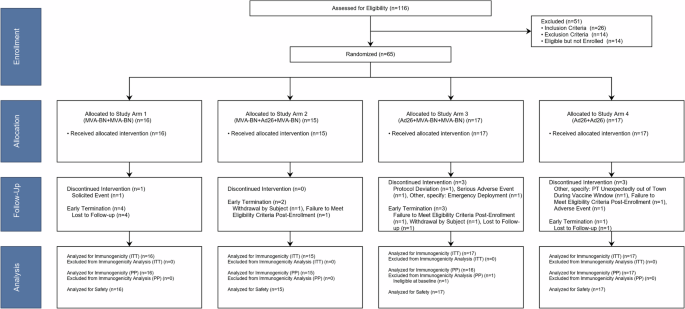
This single-center, single-blinded study was sponsored by the Office of the Surgeon General, Department of the Army, under IND 17022. The NCT Number is NCT02776761, “A Single-blind Study to Evaluate the Safety, Tolerability, and Immunogenicity of a Hantaan Puumala Virus DNA Vaccine,” registered Aug 30, 2016. Following consent and successful screening, each eligible subject was randomized at the time of enrollment into one of the three experimental groups of 9 subjects each (27 total). The study statistician randomly generated a list prior to the study and started pre-assigning specific Subject ID numbers to specific experimental groups. Then, Subject ID numbers were assigned to subjects serially based on the order of enrollment. Each subject received up to four vaccinations. Per protocol, vaccinations were administered on Days 0, 28, and 56. An optional 4th vaccination was administered on Day 168 dependent on subject availability for the additional follow-up visit on Day 196. Per protocol, subjects were followed until Day 252 (9 months). Subjects that were positive for neutralizing antibodies on Day 252 were invited to return for a day 365 follow-up visit. Subjects completed post-injection memory aids for seven days after each injection. Group 1 vaccine consisted of two administrations of 1 mg of HTNV plasmid (left and right deltoid) for a total of 2 mg/vaccination. Group 2 vaccine consisted of two administrations of 1 mg of PUUV plasmid (left, right deltoid) for a total of two mg/vaccination. Group 3 vaccine consisted of two administrations of a 1:1 mixture of HTNV and PUUV vaccines, comprised of 0.5 mg of each plasmid, 1 mg total per arm (left and right deltoid), for a total of 2 mg (1 mg of each DNA) per vaccination.
Safety assessments
The following endpoints were evaluated: (1) the nature, frequency, and severity of solicited adverse events (AEs) occurring from the time of each injection through 14 days following the procedure; (2) the nature, frequency, and severity of unsolicited AEs from the time of the first injection through 28 days following the final injection and (3) the nature, frequency, and severity of AEs from the time of the first injection through the end of the study. The solicited AEs for this study included: local findings at the site of injection (redness, swelling/induration, bruising or pain), fever, myalgias, fatigue, headache, lymphadenopathy, axillary pain/discomfort, and tachypnea. Inherent in this assessment were the medical and clinical considerations of all information surrounding the event including any medical intervention required. Each event was assigned one of the following categories: Grade 1 (mild, does not interfere with routine activities); Grade 2 (moderate, interferes with routine activities); Grade 3 (severe, unable to perform routine activities) and Grade 4 (hospitalization or ER visit for potentially life-threatening event). Laboratory AEs and abnormalities in the subject vital signs were also assessed and graded using pre-specified normal ranges within the study protocol. Safety labs included serum electrolytes, serum markers of renal (BUN/Cr) and liver function (AST/ALT), complete blood counts with differential, and urine pregnancy tests for subjects of childbearing potential.
Pseudovirion neutralization assays (PsVNA)
To assess immunogenicity, all specimens were evaluated for the presence of neutralizing antibodies using a pseudovirion neutralization assay (PsVNA)12. The PsVNA utilizes engineered vesicular stomatitis virus (VSV) that expresses a luciferase reporter gene in the place of the virus G envelope glycoprotein genes. HTNV, PUUV, SEOV, and DOBV pseudovirions (PsVs) were produced using: pWRG/HTN-M(co), pWRG/PUU-M(s2), pWRG/SEO-M(opt2), and pWRG/DOB-M, respectively13. To perform the neutralization assay, PsVs (4000 focus forming units) were combined with serum (1:20–1,562,500 dilution range) in the presence of a human complement (5%; Cedarlane, Burlington, NC, USA) and incubated overnight at 2–8 °C. The PsV plus serum mixture was then added to ATCC Vero-76 cell monolayers in clear bottom black-walled 96-well microtiter plates. The plates were incubated 18–24 h and then media removed, lysis luciferase reagent (Promega, Madison, WI, USA) added and flash luminescence data acquired using a luminometer (Tecan M200 Pro microplate reader, Mannedorf, CH). If sera contain antibodies that prevent the PsV from attaching to and/or entering cells, then the reporter activity is neutralized. Neutralization titers are interpolated from 4-parameter curves using GraphPad Prism (GraphPad, San Diego, CA, USA). The reciprocal of the interpolated dilution that results in a 50% decrease, or 80% decrease in luciferase activity is the PsVNA50, or PsVNA80 titer, respectively.
Plaque reduction neutralization test (PRNT)
Serum specimens were evaluated for neutralizing antibodies by PRNT described previously19,33. Heat-inactivated (56 C°30 min) serum samples were serial-diluted in complete media (Minimal Essential Medium with L-Glutamine [Corning] with 10% Fetal Bovine Serum [HyClone], Nonessential Amino Acids [Sigma], Penicillin/Streptomycin [HyClone], Gentamicin [Sigma], and Amphotericin B [Gibco]). An equal volume of diluted serum (antibody) was then combined with the virus diluted in complete media plus 10% human complement. This virus-antibody mixture was then incubated at 2–8 °C refrigerator overnight. The virus-antibody mixture (0.18 mL) was transferred to 6-well cell culture plates containing confluent Vero-E6 monolayers and allowed to adsorb for 1 h in a 37 °C, 5% CO2 incubator. After the absorption step, 3 mL of semi-sold overlay (EBME [Quality Biological] with 10% Fetal Bovine Serum [HyClone], Nonessential Amino Acids [Sigma], L-Glutamine, Penicillin/Streptomycin [HyClone], Gentamicin [Sigma], and Amphotericin B [Gibco], 0.6% SeaKem agarose [Lonza]) was added per well. The plates were then returned to the 37 °C, 5% CO2 incubator. For the HTNV, SEOV, and DOBV PRNT, monolayers were fixed and stained seven days after infection. PUUV monolayers were fed with 2 mL of growth medium/agarose overlay on day 7 and then fixed and stained on day 10. Fixing each monolayer was accomplished by the addition of 2 mL of 10% neutral buffered formalin per well on top of the agarose overlay, followed by 5–16 h incubation at room temperature. The agarose overlay was removed with a flat spatula, the monolayer was rinsed with PBS, and the plaques were visualized by immunostaining using horseradish peroxidase-conjugated monoclonal antibody MAb-3d7 followed by True Blue peroxidase substrate (KPL, Gaithersburg, MD, USA). PRNT50 neutralization titers were expressed as the reciprocal of the highest serum dilution resulting in at least a 50% reduction in the average number of plaques observed in the virus-only control plates.
Viruses used in PRNT
Viruses, Cells, Medium, and Antibodies. HTNV strain 76–11834, PUUV strain K2735, SEOV strain SR-1136, and DOBV strain Dobrava37, were propagated in Vero-E6 cells (Vero C1008, ATCC CRL 1586) in T-150 flasks and collected from infected-monolayer supernatants. Cells were maintained in Eagle’s minimum essential medium with Earle’s salts containing 10% fetal bovine serum (FBS), 10 mM HEPES (pH 7.4), 1x penicillin-streptomycin, amphotericin B (0.5 µg/mL) and gentamicin sulfate (50 µg/mL) at 37 °C in a 5% CO2 incubator.
Statistical methods
Descriptive analyses of safety and reactogenicity outcomes included all subjects who met the eligibility criteria, received at least one vaccination, and for whom safety data were available. Summary tables were created in which incidence, intensity, and the relationship to the use of the investigational product of individual solicited signs, symptoms, and other events were delineated by the study cohort, severity, sex, and overall. Unsolicited AEs and SAEs were analyzed in a similar fashion. For hematology and serum chemistry tests, any clinically significant change from the baseline value was identified. The median, interquartile range and normal values for each of the laboratory values (as determined by the contract laboratory) were reported for each treatment cohort for each specimen collection point. The primary analysis variable was the proportion of seropositive subjects (PsVNA50 and/or PRNT50 ≥ 20) at each scheduled timepoint for which blood samples were taken and the duration of seropositivity. Geometric mean peak PsVNA50 and PRNT50 titers were also determined for specified timepoints. Values below each assay’s limit of detection (20 for all assays) were set to 14.14 (20/√2) for analysis. Due to the geometric progression of the assay results, log10 transformations were applied to approximate normality. For all methods of comparison, transformed data were used. Agreement between PsVNA and PRNT values was analyzed using Pearson product-moment correlation. Mixed model ANOVA was used for group comparisons, with Tukey’s post-hoc tests used for specific pairwise comparisons. The Immunogenicity Population included subjects who received all three of the first three vaccinations (Days 0, 28, 56) within the acceptable visit window and attended at least one scheduled study visit subsequent to receiving the third vaccination on Day 56.
Data quality assurance
The WRAIR Clinical Trials Center conducts studies according to procedures that incorporate the ethical principles of the GCP guidelines. To ensure compliance with these procedures and to assess the adequacy of quality control procedures, the WRAIR Quality Office performed audits of the study site on behalf of the USAMRIID Quality Assurance and Regulatory Compliance Office (QARCO). Quality assurance responsibilities included visits at the initiation of the study, during the study at appropriate intervals, and after the last subject had completed the study. The WRAIR Quality Office performed the audits independently of the study monitors.


留言 (0)