
記住我





Klebsiella species are major global reservoirs and conduit of drug resistance genes between various bacterial niches [12]. We report the draft genome sequence of an ESBL-producing K. pneumoniae subsp. pneumoniae strain Cow102 previously isolated from cow milk [13, 14]. The phenotypic antimicrobial susceptibility profile of strain cow102 showed resistance to cephalosporins, carbapenem, and drugs from different classes of antibiotics including aminoglycosides (streptomycin), quinolone (ciprofloxacin, nalidixic acid), beta-lactams (amoxicillin + clavulanic acid, ampicillin, cefotaxime, ceftazidime, piperacillin), folic acid synthesis inhibitor (trimethoprim/sulfamethoxazole), monobactams (aztreonam), fosfomycin, and tetracycline. The strain was identified as an ESBL producer due to its resistance to aztreonam, cefotaxime, ceftazidime, and ceftriaxone, which are commonly used as indicators for ESBL production. The antimicrobial resistance profile and ESBL-producing status of strain Cow102 is consistent with characteristics commonly observed in ESBL-producing K. pneumoniae isolates from clinical, environmental, and animal sources in Nigeria [11, 15,16,17,18]. This suggests the pervasive nature of antibiotic resistant K. pneumoniae across different ecological niches in the country.
Haemolysis testStrain cow102 was non-hemolytic on blood agar and lacked the genes hlyA and cnf-1 that code for this function. This finding contrasts with some clinical isolates reported in various states in Nigeria [11, 15, 16, 19], suggesting potential differences in virulence mechanisms between clinical and environmental strains.
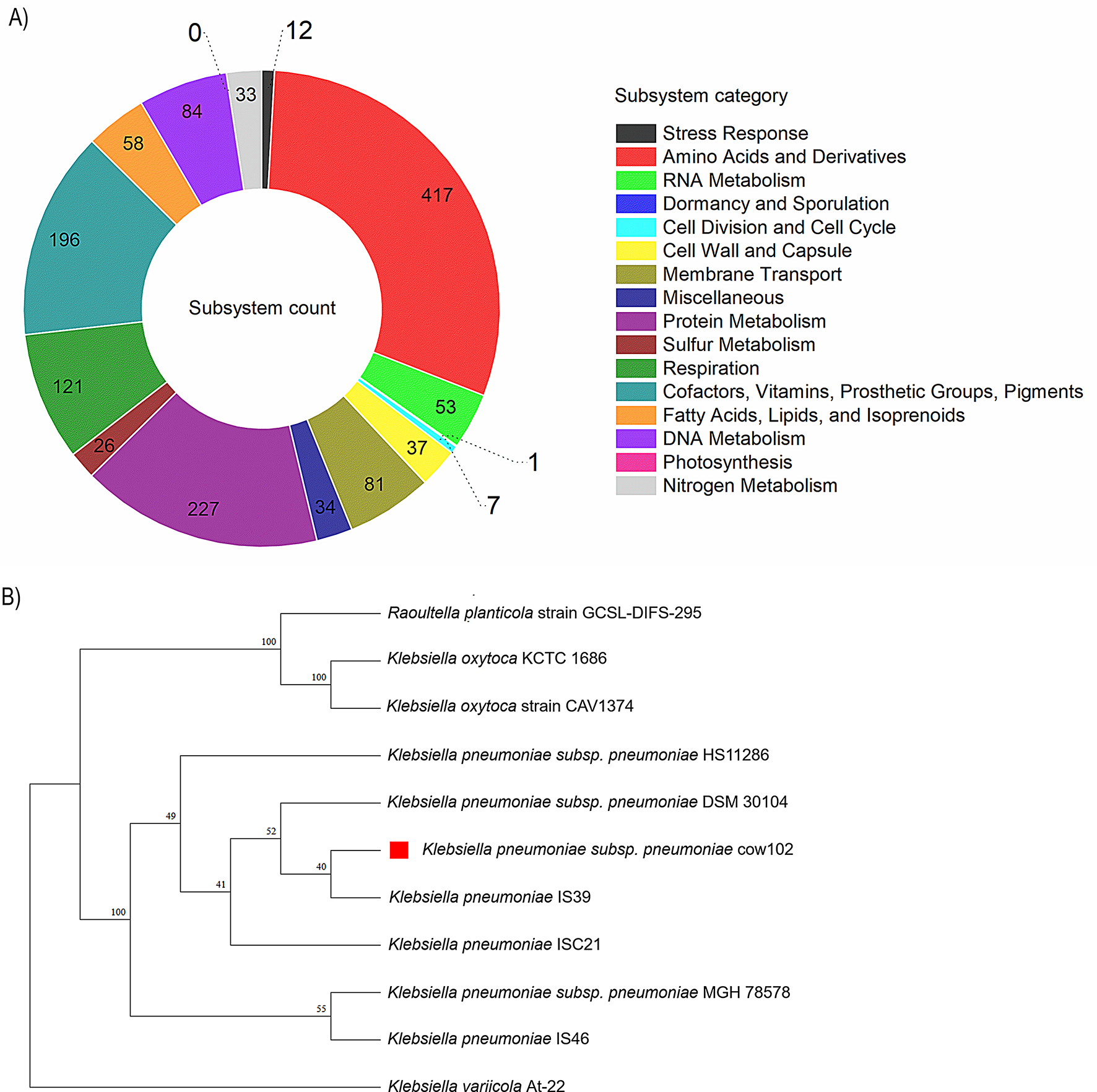
General genome featuresIllumina sequencing of the genomic DNA of strain cow102 generated 12,005,232 total read pairs (2 × 150 bp) giving approximately 336 × coverage. The assembly genome totalled 5,359,907 bp with 70 contigs, an N50 value of 376,296 bp, and a GC content of 57.35%. The assembly was characterized by CheckM as 100% complete with 0.1% contamination and had coarse and fine consistencies of 99.4% and 97.8%, respectively. Genome annotation performed using RASTk identified a total of 5,244 protein coding sequences (CDS), 72 transfer RNA (tRNA) genes, and 5 ribosomal RNA (rRNA) genes. There were 4,605 proteins with functional assignments and 639 hypothetical proteins in the annotation. Among the proteins with functional assignments, 1,457 possessed Enzyme Commission (EC) numbers, 1,208 had Gene Ontology (GO) assignments, and 1,065 contained KEGG pathway mappings. Also, about 31% of the CDS was mapped to a subsystem. Genes responsible for amino acids and derivatives (417 ORFs), protein metabolism (227 ORFs), and cofactors, vitamins, prosthetic groups, and pigments (196 ORFs) were abundant among the SEED subsystem categories [20]. An overview of the subsystem categories and feature counts is shown in Fig. 1A while the genome annotation can be viewed on Figshare [21].
Fig. 1
Subsystem distribution and phylogenetic analysis. A. Overview of the subsystems for Klebsiella pneumoniae subsp. pneumoniae strain Cow102 B. Whole genome phylogenetic tree of K. pneumoniae
Phylogenetic analysis, Multilocus sequence typing (MLST) and capsular typingThe whole-genome phylogenetic analysis showed that strain Cow102 is a sister taxon to K. pneumoniae subsp. pneumoniae strain IS39 and a close relative to ISC21, DSM30104, and HS11286 all of which are within the same monophyletic group (Fig. 1B). Monophyletic groups, also known as clades, are groups of organisms that belong to the same taxon and share a most recent common ancestor. Similar to Cow102, all related strains were reported as multidrug-resistant β-lactamase producers. Strains IS39 and ISC21 were recovered in Austria from hospital environments [22], DSM30104 was isolated in Republic of Korea from the blood of a septicaemia patient [23], and HS11286 was recovered in China from human sputum [24]. Multilocus Sequence typing analysis revealed that the current isolate had a new allelic profile [(gapA (2), infB (3), mdh (70) pgi (1) phoE (275), rpoB (44) and tonB (39)], assigned as sequence type 6914 based on the standard 7-gene MLST scheme, initially defined by Diancourt et al. [25]. eBURST analyses showed that ST6914 clustered closely with ST494, ST4138, ST4139 and ST6476 (Figure S1A). The current strain, ST6914 is a triple-locus variant to all closely clustered ST, differing at the phoE, rpoB and tonB loci. When compared with phylogenetically related strains, Cow102 shared only one or two loci with IS39 and ISC21 strains both unassigned STs, as well as with DSM30104 and HS11286, which belong to STs 3 and 11. The serotyping of ST6914 revealed a 99.96% match with K locus 122 with an unknown K type (KL122) for the capsular (K typing) antigen (Figure S1B) using the wzi sequencing K-typing method [26], and a 98.51% match with O locus O1/O2v2 with type O2afg for the lipopolysaccharide (O typing) antigen (Figure S1C).
Antimicrobial resistance genesSeveral studies in Nigeria reported drug resistant Klebsiella strains in raw animal milk and milk products; however, genomic data and serotypes of the reported strains are rare [18, 27]. Molecular characterization of the resistance factors associated with strain Cow102 revealed the presence of 38 Antibiotic Resistance Genes (ARGs) (Table 1; Fig. 2), out of which 24 genes conformed with the phenotypically derived resistance profile. The resistance genes identified in strain Cow102 include core chromosomal resistance genes belonging to the blaSHV−1 family, which confers resistance to beta-lactam antibiotics. In addition, acquired genes such as aadA2, an aminoglycoside nucleotidyltransferase gene; catA2 and catII, which confer resistance by enzymatic inactivation of amphenicol antibiotics; TEM families, which confer resistance through similar mechanisms as blaSHV−1 family; and Escherichia coli fosA and oqxA and oqxB gene conferring resistance to fosfomycin and quinolones, respectively, were found. Furthermore, the presence of the efflux pump gene, tet (D), which provides resistance to tetracycline, was expected (Table 1; Fig. 2). This is likely due to the common practice among veterinary and para-veterinary professionals in Nigeria to prescribe oxytetracycline for treating diseases [28, 29].
Fig. 2
Circular genomic map of Klebsiella pneumoniae subsp. pneumoniae strain Cow102 showing starting from the outermost ring: ring 1: insertion sequence ring 2: plasmids (coloured green), ring 3: antibiotic resistance genes (coloured red), ring 4: phage (coloured blue), ring 5: contigs from strain Cow102 (coloured red and black), ring 6: GC content (coloured black), ring 7: GC skew (coloured red and blue)
Table 1 Antimicrobial resistance genes identified in Klebsiella pneumoniae subsp. pneumoniae strain Cow102, conferring resistance to antibiotics tested using the disk diffusion methodThe genome as shown in Fig. 2 includes several additional genes associated with antimicrobial resistance that were not phenotypically tested, and their presence alone does not confirm resistance. These include katG gene and operons marA, marB, and marR, which are involved in enzymatic catalysis of antibiotics. The BcrC gene serves as a protective protein for antibiotic targets.The gdpD and pgsA genes are involved in altering the charge of the cell wall, while occD6/OprQ and oprB modulate antibiotic permeability. The presence of acrAB-tolC, emrAB-tolC, H-NS, and oxyR suggests the role of regulatory factors in modulating the expression of antibiotic resistance genes. Additionally, the genome harbours other ARGs that were acquired through horizontal gene transfer such as the E. coli uhpT gene, which carries a mutation conferring resistance to fosfomycin, mdfA gene, an efflux pump, and multidrug transporter, as well as the Haemophilus influenzae PBP3 gene which confers resistance to beta-lactam antibiotics (Fig. 2). Comprehensive details of ARGs, their orientation and genomic location is presented in Supplementary data S1.
Virulence factorsPathogens establish infections and achieve long-term survival within their host by deploying an array of virulence factors. These factors, often manifested as complex cellular structures or specialized strategies which facilitate critical functions such as host colonization, immune evasion, nutrient acquisition, and responsiveness to environmental cues [30,31,32]. However, it is important to note that the relationship between sequence type and virulence characteristics in K. pneumoniae is complex and not always straightforward. While certain sequence types may be associated with increased virulence, the presence of specific virulence genes does not necessarily correlate directly with the assigned sequence type [9, 33]. The pathogenicity of a K. pneumoniae strain is influenced by multiple factors beyond just its sequence type, including the specific combination of virulence genes, their expression levels, and host-pathogen interactions [9]. Profiling of virulence features in the current strain revealed the presence of 98 virulence factors (Supplementary Table 1) classified into 11 categories. These categories are: (i) adherence (15 genes) which allow attachment to host cells, (ii) antimicrobial activity/competitive advantage (3 genes), (iii) biofilm formation (10 genes), this is defined by the ability of microbial cells to aggregate and form a community encased in self-produced polymetric matrix [31], (iv) effector delivery system (25 genes), these are elements that facilitate the delivery of secreted effector proteins into host cells, these effector proteins alter or interfere with host processes allowing the bacteria invade, adhere, multiple and sometimes persist in the host [31], (v) exotoxins (2 genes) are secondary metabolites secreted by a microbe which interrupts or dysregulates cellular functions of the host [32], (vi) immune modulation (16 genes) allows the suppression or evasion of host immune system [30], (vii) invasion (2 genes), (viii) motility (3 genes), (ix) nutritional/metabolic factors (14 genes), (x) regulation (7 genes), and xi) other (1 gene). Hypervirulence in Klebsiella is mainly characterised by factors such as capsule type, siderophores production, lipopolysaccharide and fimbriae [34, 35]. As a result, some studies on Klebsiella focus on related genes such as mrkD, entB, iutA, rmpA, ybtS, ycfM, kfu, and allS [34,35,36]. Cow102 has an unclassified capsule serotype which indicates its distinction from the known serotypes of classical K. pneumoniae (K1 – K79), hypervirulent K. pneumoniae (K1, K2, K5, K16, K20, K54, K57, KN1) and multidrug resistant hypervirulent K. pneumoniae (K1, K2, K16, K20, K54, K62, K64 and K47) [34]. The genome of strain Cow102 contains multiple genes linked to virulence in K. pneumoniae, including mrkD, an adhesion factor that encodes the type 3 fimbrial adhesin, facilitating binding to the extracellular matrix. Additionally, siderophore systems such as entB, which is involved in synthesizing enterobactin (a catecholate siderophore), and iutA, encoding the receptor for aerobactin (a hydroxamate siderophore), are present in the genome (Supplementary Table 1). These virulence factors contribute to the strain’s ability to acquire iron (siderophores) and adhere to host surfaces (fimbrial adhesin) [34]. However, some genes commonly associated with hypervirulence in K. pneumoniae, such as rmpA, ybtS, ycfM, kfu, and allS [35], were not found in Cow102.
Mobile genetic elementsMobile genetic elements (MGEs) are enzymes or other protein encoding non-chromosomal DNA that facilitate the relocation of DNA segments either within the genome or between microorganisms [37]. Examples of MGEs include transposons (e.g., integrons, insertion sequences, gene cassettes), plasmids and phages. Figure 2 provides a visual illustration of where insertion sequences, plasmids bacteriophage are located within the CDS of the Cow102 strain. A total of 23 complete and 12 partial insertion sequences (IS) classified into ten IS families: IS1, IS3, IS5, IS6, IS21, IS110, IS66, IS91, IS630, and ISNCY in the genome of strain Cow102 (Fig. 2). A comprehensive list of all identified IS, along with their corresponding CDS positions and terminal inverted repeats (TIR) is presented in Supplementary data S2. The only plasmids found in the present genome were IncFIB(K) and Col440I. These plasmids were respectively detected in 16 (67%) and 11 (46%) of 24 K. pneumoniae isolates from powdered milk from Germany [38]. Studies have shown that the conjugative IncFIB plasmid group, known for their role in the distribution of antibiotic resistance and virulence genes, are commonly found in various strains of K. pneumoniae, contributing to their multidrug-resistant profiles [24, 39,40,41,42,43]. One such study includes a report from Brazil, where 25 out of 27 carbapenem-resistant K. pneumoniae isolates from a hospital environment were found to carry variants of the IncFIB plasmid [42]. In our study, we also observed that this plasmid was present in all the strains from Nigeria which we retrieved from the NCBI database. (Supplementary Data 3). IncFIB plasmids in Klebsiella species are crucial vectors for the transmission of ARGs and virulence genes, contributing to the pathogenicity and antimicrobial resistance of these bacteria [42, 44]. This highlights the need for further research on the genomic features and evolutionary dynamics of variants of IncFIB plasmids to combat the spread of resistance and virulence traits. Additionally, five bacteriophages were detected in Cow102, of which three (Klebsi phiKO2, Escher 500465, Entero cdtI) were intact, one (Salmon 118970) was incomplete, and one (Escher RCS47) yielded inconclusive results. The nucleotide sequence of all phagesis publicly available on Figshare. The ARGs oqxA, oqxB and an IS3 belonging to cluster 204 were traced to bacteriophage Escher 500,465. While this phage has been documented in the genomes of Klebsiella strains [36] and other enterobacteria like Pseudomonas [45] and Salmonella [46], this appears to be the first instance where it has been linked to the oqxA and oqxB genes in Klebsiella. Escher 500,465 has previously demonstrated the capacity to carry ARGs, as evidenced by its harbouring of bacA and tet(M) genes in Salmonella [46]. This finding expands our understanding of how antibiotic resistance genes can be transmitted among bacterial populations through phage-mediated gene transfer.
Comparative genomicsA comparative analysis was conducted between the genome of the current strain and nine other strains of K. pneumoniae reported in Nigeria, obtained from NCBI database. A first glance at the genomic comparison analysis suggested a high level of sequence similarity with pairwise average nucleotide identity (ANI) ranging from 99.1 to 99.9% (Figure S2). In addition, pairwise genome alignments showed high collinearity among all strains with sequence alignment percentages of about 85% and 90% (Figure S2). A closer look at the variations between the genomes using DNAdiff 1-to-1 alignment blocks found significant differences (p < 0.05) in sequence inversions, insertions/deletions (indels), relocations and translocations between the assemblies and the examined strains (Supplementary data S3). The 1-to-1 alignment blocks are specific DNA regions that can be directly and uniquely matched between two genomes or sequences being comparison. These blocks represent segments where there is a one-to-one correspondence, meaning each sequence in a reference genome has exactly one matching counterpart in the query genome, without duplications or significant rearrangements [47]. The most common structural rearrangements discovered were translocations (x̄=105), which occurred between neighbouring 1-to-1 alignment blocks in the mapping of the reference (Cow102) and query sequence (nine strains obtained from NCBI). Additionally, there was an average of approximately nine breaks in the alignment where adjacent 1-to-1 alignment blocks were in the same sequence (relocations), and two somewhat unusual breaks where adjacent 1-to-1 alignment blocks were inverted with regard to each other (inversions). Furthermore, an average of 221 insertion events, three tandem duplication insertion events (TandemIns), 32,995 Single Nucleotide Polymorphism (SNPs) and 2629 Single Nucleotide. The similarity between K. pneumoniae strains extends to their entire proteomes. Among the ten strains compared, a total of 1789 protein coding genes were predicted (Supplementary data 4). Of these, 1617 genes (90.4%) were assigned orthologous groups (OGs), while 172 genes (9.6%) remained unassigned. Orthologous genes or orthologs are groups of genes shared by different species or strains evolved from a common ancestry gene but diverged from each other because of a speciation event [49]. OrthoVenn analysis of orthologous cluster structure identified 6,299 clusters, 53,361 proteins, and 2,098 singletons (Fig. 3). Figure 3A and B display the orthologous groups in each strain, highlighting both unique and shared homologous gene clusters among them. Of these, 4,282 clusters and 43,084 proteins were shared across all strains (Fig. 3C). The ortholog analysis conducted on Cow102 revealed the presence of 4796 COGs and 5037 proteins, including 196 singletons. Additionally, three proteins were found to be unique to the strain. These unique proteins were involved in carbohydrate metabolic processes (GO:0005975), specifically phospho-cellobiase and 5-oxoprolinase subunit A3, as well as fatty acid biosynthesis (GO:0006633) through biotin-dependent acyl-coenzyme A carboxylase alpha3 subunit.
Results of comparison of the nine strains recovered from NCBI database with Cow102 revealed that 10 proteins were shared with strain T79-2, isolated from chicken faeces; 38, 9, and 1 proteins were shared with strains 157, 4502, and A117-2, respectively, isolated from blood or wounds; and 16, 15, 7, 3, and 1 proteins were shared with strains 852 K, 4595, 3600, 3264, and 1643 K, respectively, all isolated from human urine (Fig. 3). The functional enrichment analysis conducted on the shared OGs between Cow102 and the nine strains revealed the presence of strain to strain specific proteins with molecular functions. Some of these proteins that could play a role in the development or distribution of drug resistance genes include the excisionase protein (GO:0032359) with the gene ontology term “provirus excision,” shared with T79-2. This protein facilitates the excision of prophage from the host genome through site-specific recombination. Additionally, a recombination-associated protein RdgC (GO:0006310) was shared with strain 157, and a family 20 transposase (GO:0006313) was shared with strains T79-2, 4502, 852 K, and 4502. This protein is required for the transposition of an insertion element. The CRISPR system Cascade subunit CasA (GO:0043571 and GO:0051607), an adaptive immune system that provides protection against mobile genetic elements, was shared with 4502. Also present among the strain-to-strain specific proteins detected were those involved in biological processes such as the heavy metal binding protein N-acetylcysteine deacetylase (GO:0046872) shared with 4502 and 852 K. Additionally, the S-(hydroxymethyl) glutathione dehydrogenase protein (GO:0046294) involved in formaldehyde catabolic process and the S-formylglutathione hydrolase FrmB (GO:0006069) involved in ethanol oxidation were shared with strain 3600. the analysis revealed a 2,3-diketo-L-gulonate reductase (GO:0070403) protein, an NAD + binding protein, which is shared with strain 4595.
Strain 852 K had the highest number of plasmids among all the strains analysed, with a total of seven (Supplementary data S3) while strains T79-2 and 4595 were found to contain four plasmids, placing them among the top in this aspect. Only strain A117-2 shared the same number and type of plasmid, Col440II and IncFIB(K), with Cows102.
Fig. 3
Comparative genome analysis represented as (A, B) Venn diagram illustrating the distribution of shared and specific clusters of orthologous groups between Klebsiella pneumoniae subsp. pneumoniae strain Cow102 and nine type strains of K. pneumoniae reported in Nigeria. And C) occurrence table showing groups of gene clusters, including the cluster count (the number of gene clusters shared between species) and the protein count (the number of protein members in the shared clusters across these strains). Each row represents an orthologous gene cluster spanning multiple species, while each column corresponds to a different closely related strain
留言 (0)