
記住我

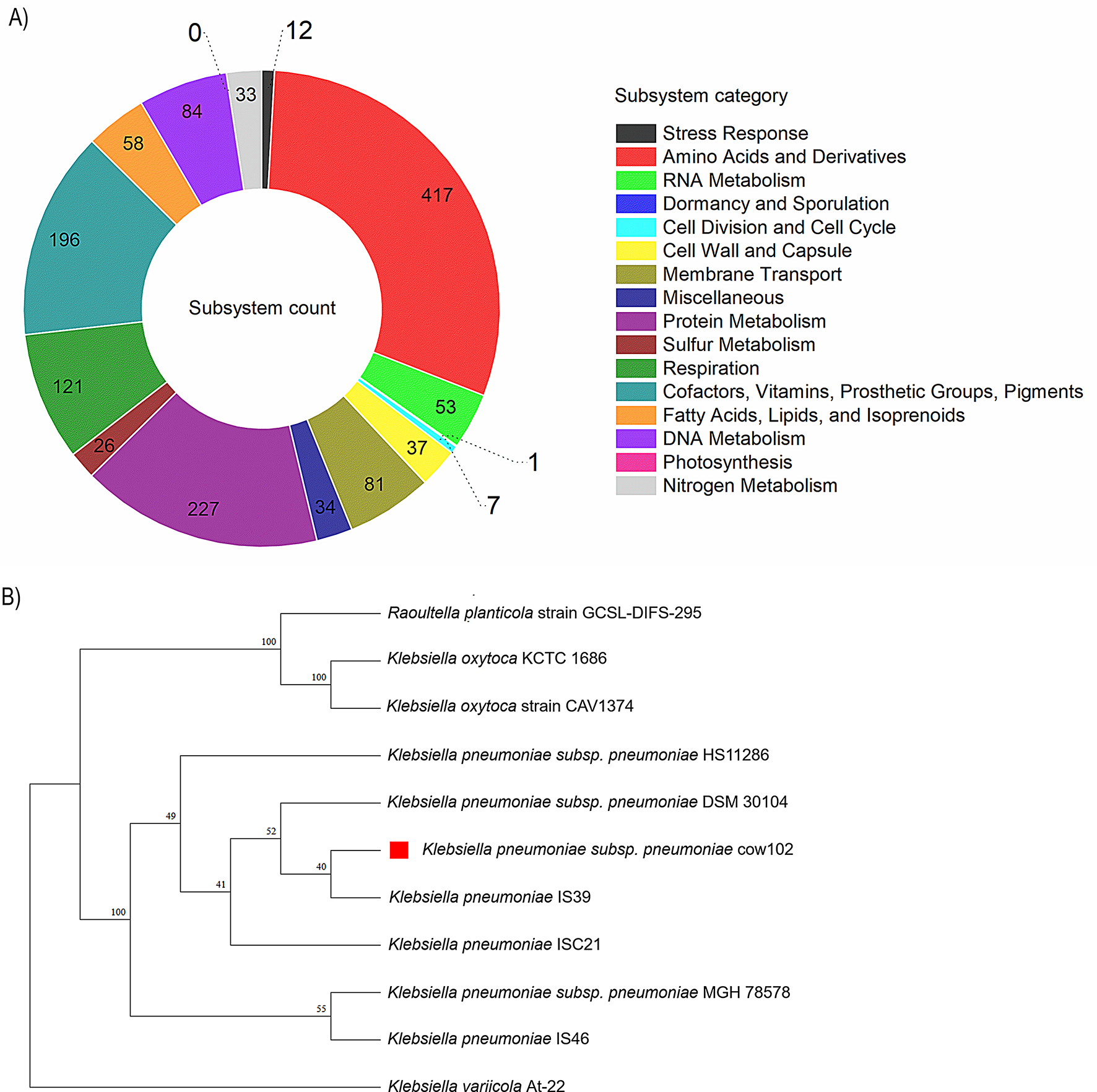
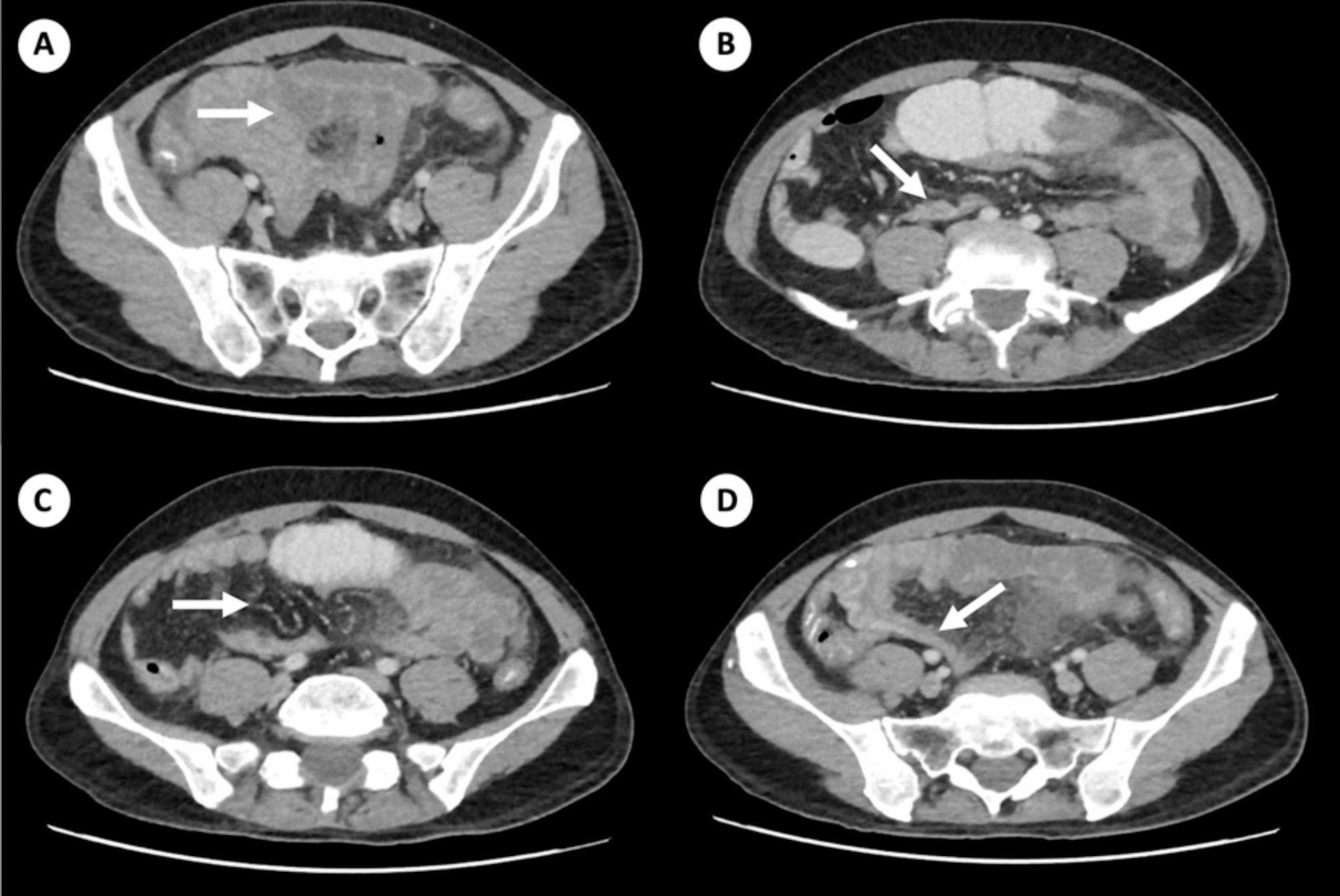




In our cohort of CD and ITB patients (n = 40; CD = 20 and ITB = 20), the mean age of the patients with CD was 42.55 ± 10.75 years, while patients with ITB had a mean age of 33.9 ± 11.80 years. Most patients with CD had ileal involvement (L1 phenotype) and stricturing B2 phenotype (50%), while the ITB patients predominantly had ileocaecal (40%) and ileocolonic (30%) involvement. The summary of the demographic and clinical details of the patient and control subjects are shown in Table 2.
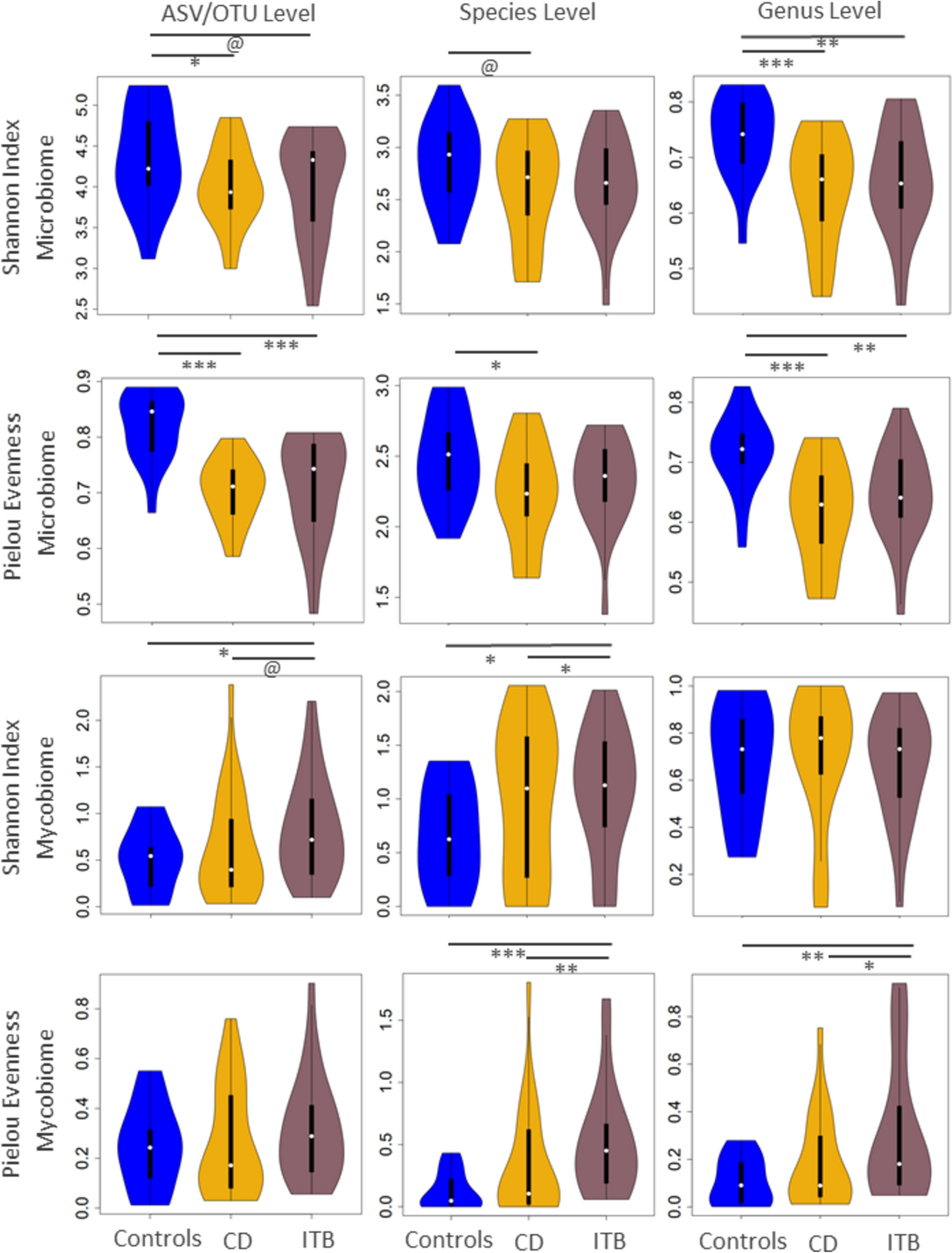
Table 2 Demographic and clinical details of healthy controls and patients with Crohn’s disease and intestinal tuberculosisBoth diseases witness differential patterns of α and β diversity variations of the gut microbiome (bacteriome/archaeome) and the gut mycobiome to controlsSubjects were categorized into three groups based on their clinical phenotypes—controls (non-CD and non-ITB individuals), and patients with CD or ITB. Both CD and ITB showed a significant reduction in gut microbiome (bacteriome/archaeome) α-diversity indices (Shannon Index and Pielou’s evenness index) when compared with controls, with evenness undergoing a more drastic reduction than the Shannon diversity metric (Fig. 1; see “Methods” section). This significant disease-associated reduction evenness was constant across three taxonomic levels (Amplicon Sequence Variants (ASVs), species and genus). Interestingly, no marked differences were observed between the bacterial α-diversities between CD and ITB (Fig. 1). The comparison of the α-diversity in the gut fungal community revealed that the ITB subjects had significantly higher Shannon Indices and Pielou’s Evenness Index when compared to the controls and CD patients across multiple taxonomic levels (Fig. 1). This indicates that both diseases are accompanied by shared alterations in the overall gut bacterial community structure, characterized by a decrease in the richness of the bacterial/archaeal members and the ITB-associated increase in the diversity and representation of the fungal members.
Fig. 1
Disease alterations in gut alpha diversity show clearly opposing patterns at the microbiome and mycobiome levels. Comparison of Shannon Index and Pielou Evenness Index for gut microbiomes and the gut mycobiomes of the three subject groups (namely Controls, CD and ITB) at the taxonomic levels of OTU/ASV, Species and Genus. Pairwise comparisons between the different groups are denoted by bars between corresponding boxes with the associated corrected p values (FDR) given in the notation: @ 0.05 < FDR < =0.1; * 0.01 < FDR < =0.05; ** 0.001 < FDR < =0.01, *** FDR < =0.001
To further investigate the community variations, we then performed a series of Principal Coordinate Analyses (PCoAs) to find the variation patterns in the overall gut bacteriome/archaeome and mycobiome compositions across the three groups (see “Methods” section; Supplementary Figure 1 for a pictorial summary). The PCoA plots at the gut microbiome level (bacteriome/archaeome) depicted a marked overlap between the CD and ITB samples, while the controls clustered separately from both diseases with significant variations noted across the first PCoA axes (PCo1) (Fig. 2A, B). At the gut mycobiome level, however, the PCoA highlighted significant variations between the control and ITB samples, indicating a distinct make-up of the gut fungal community in the ITB patients, while the CD patients showed intermediate positioning between the controls and ITB patients (Fig. 2A, B). The pattern was consistent across all taxonomic levels and distance matrices (Supplementary Figures 2–6; Supplementary Table 1). The α and β-diversity analysis indicates that both diseases undergo shared microbial dysbiosis but with distinct disease-specific patterns emerging for the bacteriome/archeaome and fungal components.
Fig. 2
ITB and CD indicate distinct patterns of microbiome- and mycobiome-level alterations with respect to the control group. A Principal Coordinate Analysis (PCoA) plots showing the variations of the Species-level Gut Microbiome profiles and Gut Mycobiome profiles of individuals belonging to the Control, ITB and CD groups. The PCoAs were performed using the weighted Jaccard distance measure. Species-level PCoAs obtained for the Kendall distance measure have been shown in Supplementary Figure 1. Similar plots for the other taxonomic levels using other distance measures are provided in Supplementary Figures 2, 3, 4, and 5. B Boxplot comparing the first species-level PCoA Axes obtained for the gut microbiome and mycobiome profiles obtained using the three different measures. Pairwise comparisons between the different groups are denoted by bars between corresponding boxes with the associated corrected p values (FDR) given in the notation: @ 0.05 < FDR < =0.1; * 0.01 < FDR < =0.05; ** 0.001 < FDR < =0.01, *** FDR < =0.001
Specific gut microbial members diagnostically distinguish disease-associated dysbiosis and enable the distinction between CD and ITBNext, we identified the taxa (or modules of specific taxa) that distinguished between CD and ITB and between the two diseases and controls, and the extent to which they contributed to the distinction. For this, we adopted the machine learning-based Random Forest classification approach, which not only identified the most discriminatory markers for pairwise group classification using the entire dataset but also validated these markers using an iterative bootstrapped approach to preclude biases originating from outlier samples (see “Methods” section for a complete description of the methodology; Supplementary Figure 7 for a complete pictorial description). The diagnostic markers were assessed separately for the gut microbiome and mycobiome.
At the microbiome level, we identified the most discriminatory taxonomic features (at genus and species level) achieving the highest diagnostic accuracy for pairwise discrimination between Controls vs. ITB (70 features; top AUC: 94.5%), Controls vs. CD (50 features; top AUC: 98.5%) and CD vs. ITB (10 features; top AUC: 76%), respectively (Fig. 3A, B). This indicated that while the microbiome alterations in the two gut inflammatory diseases were distinct from the controls, the disease groups also harbored subtle yet specific variations in their gut microbial community makeup. A similar pattern was also observed for the gut mycobiome community. We identified 10 fungal features enabling distinction between the controls and ITB group, and 30 fungal features discriminating the control vs. CD groups and the CD vs. ITB groups. The mycobiome alterations had the best capability to distinguish between Controls vs. ITB (94.4% AUC), followed by Controls vs. CD (83.3% AUC) and CD vs. ITB (75.7%), reflecting the trends observed for the gut microbiome. These observations highlight that the gut bacteriome/archaeome and mycobiome profiles couldn’t just reliably distinguish between the patients (CD/ITB) and controls but also distinguished the diseases with appreciable accuracy. This entire investigation identified a total of 85 gut bacteria and 37 gut fungal taxa at the species and genus level that could facilitate diagnostically distinguishing between at least one pair of the three sample groups. These taxa have been summarized in Fig. 3C and D.
Fig. 3
Identification of differentially associating taxonomic features that are diagnostic of the three different groups (Controls, CD and ITB). A Identification of the most diagnostic features for each pairwise group classification, using multiple Random Forest models each considering only a varying number of top features (e.g. top 10, 20, 30, 40, till 250). For each pair of groups (Controls vs. CD, Controls vs. ITB, CD vs. ITB), the most diagnostic set of features were the ones corresponding to the model with the highest classification AUC. B Boxplots comparing the AUC ranges for the 50 iterative bootstrapped Random Forest (RF) model variants. As shown, the variants were generated for discriminating between each pair of groups (Controls vs. CD, Controls vs. ITB, CD vs. ITB), each considering only the top features identified (in A) for corresponding group-pair. For a given pair of groups, to create the RF variant in each iteration we randomly selected 50% of the samples (for generating the training RF model). This model was then tested on the rest 50% of the samples (corresponding to the concerned pair of groups). C–D Heatmaps showing the cross-group variation of the different taxonomic features identified in A to be amongst the top features discriminating across at least one pair of groups at the microbiome (i.e. Bacteriome/Archaeome) (C) and the mycobiome level (D). The upper heatmap groups these features based on their differential abundance/detection across each of the subject group pairs (Controls, ITB vs. Controls, ITB vs. CD; indicated by green color in the lower heatmap). In this scenario, for any pair in the notation ‘A vs. B’, the taxonomic features increased (in abundance or detection) in A with respect to B are highlighted in different shades of pink (FDR < =0.1) and those that are decreased are denoted in different shades of blue as denoted by the key. Markers that enable discrimination between controls vs. ITB/CD are highlighted in boxes with yellow boundaries, while those that facilitate distinguishing between ITB and CD are shown in boxes with blue lines. Abundance of features are denoted in blue font and detection are denoted in blue fonts
The microbiome taxa that could distinguish between controls and the two diseases and were enriched in the diseased group to controls included Faecalibacterium (F. prausnitzii), Roseburia (R. inulinivorans), Coprococcus (C. catus), Eubacterium (E. ramulus, E. rectale, E. eligens, E. desmolans), Dorea (D. formicigenerans, D. longicatena), Catenibacterium, Finegoldia (F. magna), Blautia (B. faecis, B. luti), Prevotella, Megamonas (M. funiformis) (fig. 3C). At the mycobiome level, the taxa with the same pattern of abundance alterations included Blumeria, Aspergillus penicillioides and Alternaria tenuissima. On the other hand, the diagnostic taxa that were observed to be increased in both diseases with respect to (w.r.t) controls included Fusobacterium, Hydrotalea (H. flava), Streptococcus (S. thermophilus), Collinsella, Gemella (G. haemolysans), Shewanella (S. amazonensis), Methylobacterium (M. aquaticum), Parabacteroides (P. distasonis), Veillonella (V. dispar, V. parvula) at the microbiome level and; Fusarium, Saccharomyces (S. cerevesiae), Parengyondontium (P. album) and Xenoacremonium (X. falcatum) at the mycobiome level (fig. 3D).
At the bacteriome level, while Bifidobacterium and Finegoldia (F. magna) were specifically enriched in ITB w.r.t CD, Bilophila showed the opposite trend (enriched in CD w.r.t ITB). Additionally, the pathobiont Ruminococcus gnavus, along with Lactobacillus (L. salivarius), Solobacterium moorei, Bacteroides stercoris, Collinsella (C. aerofaciens) were significantly enriched in ITB w.r.t Controls (but unaltered in CD vs. controls analysis). Similarly, at the mycobiome level, Candida tropicalis, Alternaria metachromatica and Phanerochaete sp. showed significant enrichment in ITB w.r.t CD. Additionally, the overall abundances of Malassezia, Candida, and Alternaria showed a significant increase in ITB (w.r.t control) and a trend of enhancement in CD.
Differential taxa can be grouped into three distinct co-abundant modules in the gut microbiomeNext, we investigated if these diagnostic taxa could be arranged into specific co-abundant modules. For this purpose, we generated a co-abundance network of the above-identified 122 taxonomic features (85 gut bacterial and 37 gut fungal) (see “Methods” section). Separate co-abundance networks were constructed for the gut microbiome and mycobiome profiles.
The co-abundance network depicting the associations between the differentially abundant taxa consisted of three distinct modules. The “Disease Depleted” module consisted of putative beneficial bacterial taxa that were depleted across both diseases (highlighted in the previous section), such as Faecalibacterium, Roseburia inulinivorans, Eubacterium eligens, Coprococcus catus, Coprococcus comes, Blautia luti, Dorea longicatena, Eubacterium ramulus, etc. (Fig. 4A, B). Taxa that were enriched across both diseases, as identified in the previous section (Fig. 3C, D), comprised lineages like Shewanella, Hydrotalea, Methylobacterium, etc. (Fig. 4A, B). Amongst the ‘disease-enriched’ module, only Bifidobacterium was found to be enriched in ITB when compared to CD. Two sub-modules were also observed, one comprising Finegoldia, which increased in ITB compared to CD, and the other containing Bilophila (B. wadsworthia) enriched in CD compared to ITB (Fig. 4C).
Fig. 4
Co-abundance network of diagnostic taxonomic features highlighting the three different modules. The three images in A–C show the same network, highlighting the only the differentially abundant taxa observed for A ITB vs. Controls, B CD vs. Controls, C ITB vs. CD. In this scenario, for any pair in the notation ‘A vs. B’, the taxonomic features increased (in abundance or detection) in A with respect to B are highlighted in pink (FDR < =0.1) and those that are decreased are denoted in blue as denoted by the key
The mycobiome co-abundance network was observed to consist of a central hub of multiple taxa linked through Parengyodontium to a Candida-specific sub-hub. While both the major hub and the sub-hub were observed to be increased in ITB vs. Controls, CD was enriched only for the major mycobiome hub to controls. There were also multiple disease-specific alterations within the mycobiome network. While Candida tropicalis was observed to be enriched in ITB vs. CD, Alternaria metachromatica and Phanerochaete showed the opposite trend.
To check the reproducibility of our findings on the co-abundance associations, we validated our module-specific co-abundance relationships using the global gut bacteriome/archaeome and mycobiome datasets. Identification of disease-associated modules reproduced in multiple global cohorts could indicate universal gut bacteriome/archaeome and mycobiome signatures which could be exploited as microbiome-based diagnostics and/or therapeutics. To evaluate this aspect, we developed intra-bacteriome and intra-mycobiome networks encompassing >5400 gut bacteriome profiles and ~900 gut mycobiome profiles collated from 14 studies across the globe (Table 1; Supplementary Figures 8, 9). We then investigated the reproducibility of the intra-modular edges observed within each module (Fig. 4), in the meta-networks identified in Supplementary Figures 8 and 9.
The intra-mycobiome associations showed distinctively low reproducibility across global cohorts. With threshold q value < =0.1, we could observe links between specific genera and their respective species (as expected) but no cross-genera associations. Relaxing the threshold to p value < =0.05 increased the number of associations to 33 with only a few cross-clade links (Malassezia restricta—Penicillium citrinum; Blumeria—Malassezia; Blumeria—Trichosporon asahii; Trichosporon—Aspergillus). These results indicate the likelihood of intra-mycobiome associations being cohort-specific with low reproducibility across cohorts (Supplementary Table 2; Supplementary Figure 9).
In contrast, the gut microbiome meta-networks revealed distinct patterns in the positioning of the taxa belonging to the ‘Disease-Depleted’ and ‘Disease-Enriched’ microbiome modules (Supplementary Figure 8A). Taxa belonging to the ‘Disease-Depleted’ module were observed to occupy central positions in the cross-cohort meta-network, showing significantly higher degree centrality measures (Mann–Whitney p value < =0.002; Supplementary Figure 8A, B). We also observed stark differences in the reproducibility of these intra-modular edges across the different modules in the global meta-network with the intra-microbiome co-abundance network of taxa depicted in Fig. 4. Amongst the reproducible edges across the two co-abundance networks, more than 65% belonged to the ‘Disease-Depleted’ module.
This indicated that while the associations within the ‘Disease-Enriched’ and ‘Mycobiome’ modules are sporadic and observed only in the specific cohort of this study (as well as a likely consequence of indirect associations occurring due to other gut ecological changes), the taxa belonging ‘Disease-Depleted’ module not only constitute the central core of the global gut microbial community, but associations within this module are reasonably conserved across cohorts. This indicates a specific ecological role for these ‘disease-depleted’ taxa as ‘cornerstones’ within the microbiome whose depletion is linked with the onset of both diseases.
Loss of core gut microbiome is linked with the gut inflammation phenotype globally across multiple studiesPrevious studies have indicated the loss of stability of the microbiome in IBD, with multiple studies including the current one, reporting the loss of diversity and diminishment of certain microbial taxa in gut inflammatory disorders [48,49,50,51]. Thus, we next checked if the two “Disease-Depleted” modules encompassed members of a core, health-associated gut microbiome, whose loss was driven by the inflammatory phenotype. Our hypothesis herein was that the “health-associated” microbiome module contained specific members that were not only prevalent across cohorts (as observed from their association with health and reproducible presence in global cohorts), but also influenced the stability of the microbiome (which could be a consequence of these taxa occupying the central nodes of the microbiome). To investigate this, we attempted to identify the core of the Indian gut microbiome. We collated 1617 gut microbiomes from seven studies. Subsequently, using a combination of prevalence and meta-network degree centrality (see “Methods” section), we identified a list of 46 taxa (28 species and 18 genera) that were prevalent (Detection in at least four of the seven datasets; 70%) and central (degree centrality in the top 70 percentile) in the Indian gut meta-network (derived from the seven studies) (Fig. 5A, B; Supplementary Figure 10). More than 75% of the Indian core gut taxa belonged to the ‘Disease-Depleted’ Module-1 indicating that both the diseases were associated with the loss of this centrally connected core module in the gut microbial community.
Fig. 5
Loss of the core gut microbiome is associated with gut inflammation and its severity across multiple studies. A Multi-study meta-analysis approach utilized for identification of the core gut microbiota in the Indian populations. B Co-abundance meta-network shown only for the core taxa in the India gut microbiomes derived from the seven studies. C Degree centrality of the identified core India gut taxa at the species level. Taxa are listed in descending order of their degree of centrality. D Comparison of the CGMS in different inflammation disease phenotypes in the current study, and the previous Kedia et al. study from India [13, 39]. The CGMS are also compared for five additional external studies from North America [8, 45,46,47], two studies from Europe (Liguori et al. from Italy [32]; Halfvarson et al. from Sweden [46]). E. Heatmap showing the association of four major indices of gut health along with CGMS with IBD-disease groups across the seven cohorts. Red boxes marked −1 denote a significant decrease (with Mann–Whitney test p value < =0.05); blue boxes marked 1 denote a significant increase (with Mann–Whitney p value < =0.05). Depicting the comparative evaluation for their association with non-diseased and IBD-patients across all investigated cohorts
Next, we probed if this loss of the core gut microbiome could be utilized to profile the extent of gut inflammation. We devised a simple quantitative score called the core gut microbiome score (CGMS). The CGMS for the microbiomes belonging to a given dataset was the summation of the rank-scaled abundances of the core Indian gut species (Fig. 5C) that were detected in the given dataset, with the abundances of each species being weighted by its percentile in the degree centrality in the meta-network (Fig. 5A).
We then investigated seven study cohorts (including the current study) from India, Sweden, the US and Italy and investigated the variation of the CGMS across different inflammation phenotypes. In the current study, the controls were observed to have significantly higher CGMS values as compared to CD and ITB (with no significant differences between the two disease types) (Mann–Whitney FDR < =0.05). On the other hand, our previous study investigated two different inflammatory disorders (CD/UC and Acute Severe Ulcerative Colitis: ASUC) [23]. ASUC subjects have a higher degree of inflammation than CD/UC. This was also reflected in the CGMS scores, wherein the controls had CGMS values significantly higher than either CD/UC or ASUC (Mann–Whitney FDR < =0.001 for both), and ASUC had significantly lower CGMS values compared to CD/UC (Mann–Whitney FDR < =0.001), indicating a greater loss of core in ASUC.
We next investigated if the CGMS scores defined on the Indian population could predict inflammation phenotypes in geographically distinct datasets. For this purpose, we investigated the IBD cohort of the Human Microbiome Project [45]. Although defined on the gut microbiome core of the Indian population, the CGMS scores could efficiently distinguish between Controls and IBD patients with the CGMS for patients being significantly lower (Mann–Whitney p value < =0.001). We also investigated another Italian cohort [32], which had gut microbiome profiles sampled from Controls, and IBD patients with active disease and those in remission. Even in this case, we observed that the CGMS values of the controls were significantly higher than those of the patients. The values of individuals in remission were noticeably higher than the patients, indicating a partial core recovery. Two additional cohorts [Hall et al. (n = 259) and Lloyd-Price et al. (n = 1627)] [8, 45] consisting of gut microbiome profiles of healthy controls and of the patients with IBD, showed significantly low CGMS in the diseased samples when compared with controls. Swedish cohort [Halfvarson et al. (n = 683)] too, showed a significant reduction in CGMS in CD [46]. Thus, these results indicate that the abundance profiles of the core members of the microbiome can be utilized to measure the inflammation status of individuals.
We then compared the efficacy of the CGMS (as an indicator of gut health) with four other indices previously proposed for this purpose. These included the Shannon Diversity and the GMHI (both known to be positively associated with controls or ‘healthy’ individuals), and Dysbiosis-Score and Kendall Uniqueness (known to be associated with diseased groups) This comparative evaluation showed CGMS as the only indicator showing significant reduction in the IBD-disease groups as compared to the controls across all seven cohorts. The GMHI and Dysbiosis-Score were second in performance showing significant decrease and increase in five out of the seven cohorts respectively, followed by Kendall Uniqueness (significantly increased in four cohorts). The worst performing indicator was Shannon Diversity (Fig. 5E). This indicates that the CGMS is able to perform equally or better than other indicators of gut health in the seven cohorts.
Investigation of taxa-inferred metabolic profiles identifies specific metabolites whose production or consumption/degradation associated with loss of control-associated microbiomeIn the last part of this investigation, we inferred the putative metabolic functionalities in the different gut microbiomes based on their taxonomic composition. For this purpose, we utilized taxa-to-metabolic functionality maps of experimentally validated metabolic functional profiles collated and used as part of multiple previous studies (See Methods). Given the species-level-taxonomic composition of a gut microbiome, this approach enabled inferring of metabolic profile given the abundances of its constituent taxa (see “Methods” section).
Comparing the inferred metabolic profiles (using Principal Coordinate Analysis or PCoA) across the three groups of subjects included in this study identified significant differences across the three groups (PERMANOVA using Bray–Curtis: R-squared = 0.11 and p = 0.009) (Fig. 6A), with functional variations across the three groups, most prominent along the first Principal Coordinate PCo1 (Fig. 6B). The most significant variation of the functional profiles was observed between the ITB and the Control subjects (Mann–Whitney p value = 0.005), with the CD group at intermediate position.
Fig. 6
Identification of metabolites associated with the progression from controls towards CD and ITB in terms of consumption, degradation and production through inferred metabolite profiling. A The Principal Co-ordinate Analysis (PCoA) plot describes the variation in microbiomes in three different groups (Controls, CD and ITB) which has been shown in different color legends. Bray–Curtis distance measure has been used to perform the PCoA analysis and the p value and R2 value obtained from PERMANOVA using Bray–Curtis distance has been mentioned as well. B The top right plot (box plots) captures the values of Principle Co-ordinate 1 across three different groups (Controls, CD and ITB) and the significance in the difference between groups have been shown through the p value calculated through Wilcoxon Rank-Sum test. C Volcano plot showing the association of different metabolite production profiles with PCo1 (p values obtained from the Spearman correlation analysis were adjusted using Benjamini–Hochberg method for multiple corrections). The same volcano-plot for the consumption/degradation profiles is shown in D. The directionality of association of different functionalities with PCo1 as well as with Control or Diseased microbiome is also indicated. Functionalities that were also validated in the Random Effect Model meta-analysis in Supplementary Figure 11 are shown in red-boxes. E Heatmap on the left panel shows the different taxa associated with the different BA production/consumption profiles. The bar-plot in the middle shows the association of these taxa with PCo1 values of part A of this figure (positive values indicating disease association). The right most heatmap shows the enrichment or depletion in IBD patients across the global cohorts
We further identified the different metabolic functionalities whose abundances associated with Control-to-Disease variation along the PCo1, by correlating the abundance of each metabolic functionality (i.e. the cumulated abundance of the taxa having that metabolic functionality) with PCo1 values. We identified a total of 43 metabolite production profiles significantly associated with PCo1 values. While the production of all three short-chain fatty acids (Acetate, Propanoate and Butyrate), along with other metabolites like CO2 and Methylamine, were putatively enriched in controls, the metabolite production profiles associated with disease-states contained, besides other, the production of the two primary bile acids (BAs), cholic and chenodeoxycholic acids and the secondary hydrophobic BAs (deoxycholic and lithocholic acids) (Fig. 6C). We also identified 83 metabolic consumption profiles associated with PCo1 values. Notably, the list of most positively associated metabolite consumptions included the consumption of glycine- and taurine-conjugated BAs (Fig. 6D). This indicated that diseased microbiomes in our study were enriched for taxa consuming glycine- and taurine-conjugated BAs and producing primary and secondary BAs.
Since the analysis involved inferred metabolic functions, we further checked if the enrichment of these functionalities with disease were also replicated in global cohorts. We thus computed inferred metabolite profiles across six other global matched IBD-control cohorts (investigated in Fig. 5D, E) and performed a Random-Effect Model meta-analysis. Validating the associations, the positive association of taurine-conjugated BA consumption and an increased production potential of primary BA and secondary BA (de-oxycholic: DCA) with IBD phenotypes was replicated in this global meta-analysis (Fig. 6C, D; Supplementary Figure 11).
Thus, we next checked which specific microbiome members were associated with these specific functions in our study cohort. This revealed that the above conversion functionalities were primarily present in multiple Bifidobacterium and Lactobacilli, along with specific Bacteroides species (fragilis/vulgatus/thetaiotamicron) (Fig. 6E). Amongst them, six taxa, Bifidobacterium longum/breve, Lactobacillus gasseri/johnsonii and Bacteroides fragilis/vulgatus were observed to be significantly enriched in disease microbiomes. We further validated these associations across the global cohorts. Despite cohort-specific variations, despite being known probiotics, B. longum, B. breve, and L. gasseri, were significantly enriched in IBD patients across multiple cohorts, along with B. thetaiotamicron, indicating that these alterations are replicable in other cohorts.
留言 (0)