
記住我

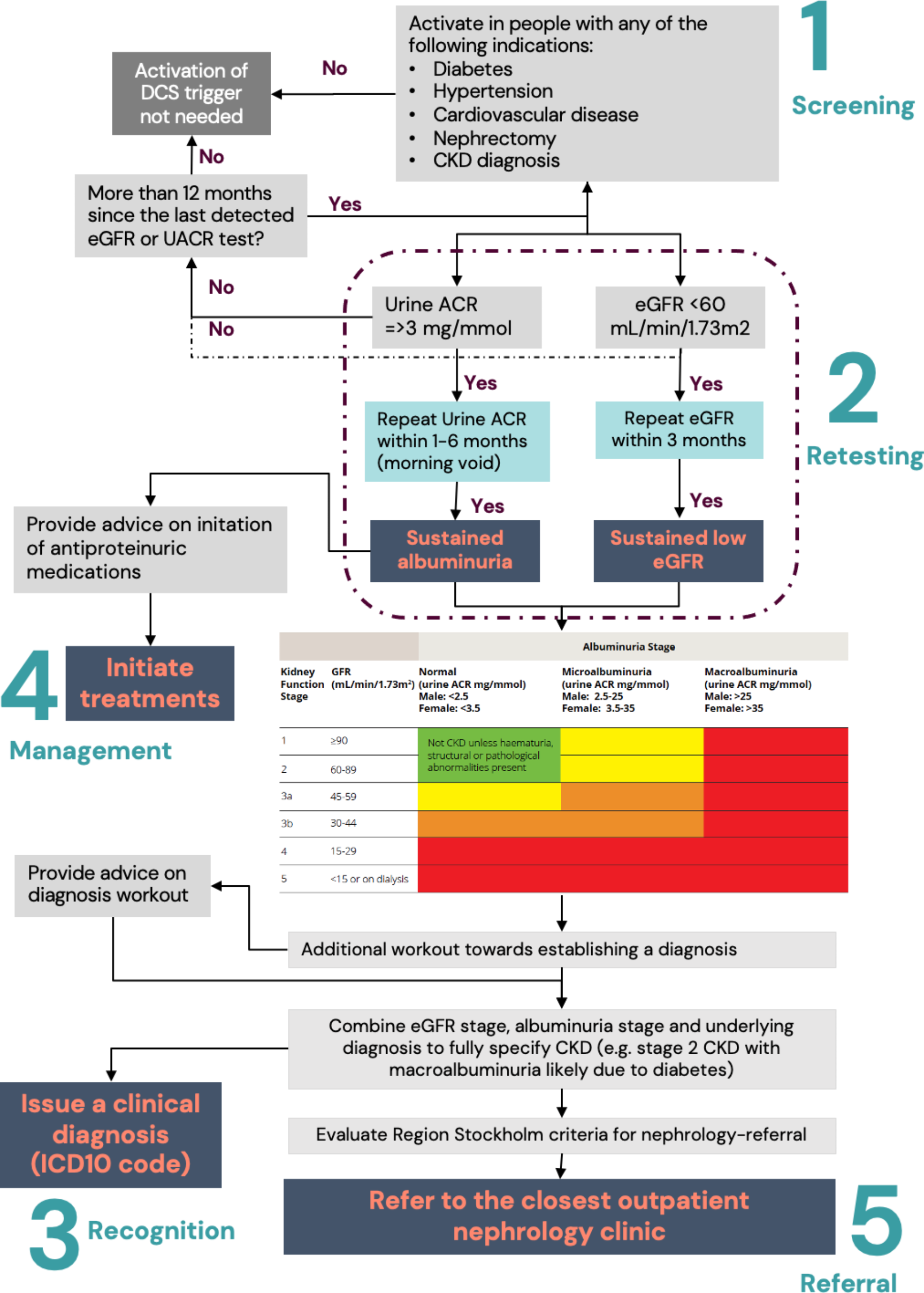




We translated the regional [7] and national [6] guidelines for CKD identification and management in primary care into ALMA-triggers and facilitators. This resulted in a battery of CDS triggers grouped into 5 different processes of CKD care (Fig. 1). Selected key examples of how these triggers operate are described in Table 1.
Fig. 1
Schematic representation of the five main steps considered for the identification and management of CKD in primary care
Table 1 Selected examples of CDS triggers and advice/practice facilitators offered to the physicianThe CDS triggers were developed by a multidisciplinary group of nephrologists, primary care physicians and software developers. Software engineers at ALMA developed the CDS triggering algorithms that were internally tested in an artificial environment. As a next step, the CDS triggers were tested in a pilot phase at Gustavsberg academic primary healthcare center. Physicians at that center experienced the CDS triggers during a 2-week period, provided feedback on its functionality and gave suggestions for improvement of it. We then determined that the CDS-triggering system was ready for evaluation in a clinical trial.
Trial designThis is a pragmatic cluster-randomized, parallel, two-arm controlled clinical trial (ClinicalTrials.gov ID NCT06386172) involving all public non-subsidized primary care centers in the region of Stockholm (n = 66) with the goal to compare the usefulness of ALMA-CKD triggers versus the routinely issued CKD advice currently provided in the ALMA platform. Retrospective registration of the trial protocol was first posted on clinicaltrials.gov on 2024-04-26. The trial sponsor is Karolinska Institutet.
Trial armsThe 66 primary care centers that used the ALMA platform were randomized in proportion 1:1 to:
Control arm: Be exposed to the advice/support already-available in the ALMA platform regarding management of kidney disease that consists of: a)Recommendation to test for albuminuria once annually in patients with diabetes; b)Recommendation to start ACE-inhibitors in patients with diabetes and albuminuria; c)Recommendation to establish a diagnosis of diabetic nephropathy in patients with diabetes and eGFR < 60 ml/min/1.73 m [2] or albuminuria; and d) alert if there is a recorded decrease of eGFR of more than 20% in the last 18 months.
Intervention arm: In addition to the advice already provided by the ALMA platform, be exposed to additional advice/support related to the management of CKD (ALMA-CKD CDS triggering system).
Clinicians working at the healthcare centers will be blinded as to which arm the center has been allocated. The research team will be blinded, other than members who work for Blackwell Medtech and that by necessity must implement the intervention. There are no plans to unblind center staff or the research team.
The randomization procedure is described below.
Study endpointsStudy outcomes are defined at the level of individual patients.
The primary outcomes are creatinine and albuminuria screening within 12 months among those with an indication. For clarity, for each patient visiting one of the participating centers, and who meets the indication for annual kidney function screening, we will evaluate and compare whether they received a screening test for these biomarkers. We will evaluate the testing of each filtration marker separately. Patients eligible for screening are, according to current guidelines, those with a diagnosis of CKD, hypertension, diabetes, cardiovascular disease or nephrectomy.
The secondary outcomes refer to subsequent processes of CKD care as follows: For each patient visiting one of the participating centers, who meets the indication for re-testing of creatinine or albuminuria, we will measure whether a re-test was performed within six months of their original test date. An indication for re-testing creatinine is having eGFR < 60 ml/min/1.73 m [2] in the original test, and an indication for re-testing albuminuria is having a dipstick test denoting KDIGO A2 + or a urinary albumin to creatinine (uACR) test > 3 mg/mmol in the original test. This analysis will exclude those participants carrying already a clinical diagnosis of CKD.
For each patient visiting one of the participating centers, who continue to have eGFR < 60 ml/min/1.73 m [2] and/or dipstick A2 + or > 3 mg/mmol at re-testing, we will measure the proportion of issued clinical diagnoses of CKD (by ICD-10th system) within 6 months. This analysis will exclude those participants carrying already a clinical diagnosis of CKD.
For each patient visiting one of the participating centers, who continue to have dipstick A2 + or > 3 mg/mmol at re-testing, we will measure the proportion of patients who start CKD-modifying agents (ACE/ARBs, SGLT2-i, etc.) or, if a CKD-modifying agent was already being dispensed, whether a second line of therapy was started, both within 6 months.
For each patients fulfilling criteria for referral to nephrologist care, we will record the proportion of referrals for ultrasound of the kidneys and the proportion of accepted referrals to nephrologist specialist care within the intervention period. Criteria for referral to nephrologist care are those recommended by Swedish guidelines [6]. This analysis will exclude those participants with already recorded visits to the nephrologist in the 2 years prior to trial start.
Analysis population and trial durationAs this is a pragmatic trial, all analysis will be performed on an intention to treat population, consisting of all eligible patients, analyzed according to the randomized allocation of the healthcare center. Each individual will only be included in each analysis once; that is, they will be included into the analysis population for each endpoint on the first time they visit a participating center during the recruitment phase and meet the eligibility criteria. If they do not meet the criteria on the first visit, but do on a subsequent visit, they will be included on that second visit, but the same individual will never be included twice in the same analysis set.
All patients who visit a participating healthcare center at some point during the 12 months following randomization will be considered for inclusion, and all will be followed up for a further 12 months. Hence the trial will run for a total of 24 months.
Activities to enhance use of algorithmsTwo initiatives will be implemented to increase awareness of the ALMA-CKD triggers amongst the general practitioners at the centers allocated to the intervention arm. Within 3 months from trial start, an informative video on the new ALMA-CKD features was sent to all attending clinicians and posted in the internal website of the SLSO centers (but only visible by the people employed in the centers randomized to intervention). Within 6 months from trial start, 2 more specific educational videos on how to benefit from ALMA-CKD will be posted in the educational section of the SLSO internal website (again, only visible by the people employed in the centers randomized to intervention) and sent to the managers of respective center for distribution.
Pragmatic trial with no active data collectionThis is a pragmatic trial, and as such we have designed it to minimize interactions between patients and researchers, so as to avoid interfering with routine of care. Patients will receive care according to the decisions of their clinician in their choice of healthcare center and will not be asked to make additional visits or provide additional samples for the purposes of this study.
A novelty of this trial is its embedment in the well-established Stockholm CREAtinine Measurements (SCREAM) data repository [20]). By this feature, the study design resembles a registry-randomized RCT, which is a modern and effective way to run pragmatic trials, benefiting from the excellent information collected in Swedish databases, and minimizing trial costs as there is no need to call participants for programmed visits, checkups or samplings. As such we will not have a Data Monitoring Committee.
SCREAM is a complex linkage of administrative and clinical healthcare databases from Region Stockholm with continuous updates since 2006 [20]. For this trial, the following SCREAM databases needed are:
1.The administrative health data registry of Region Stockholm (Vård Analys Databasen, VAL; the Region Stockholm healthcare data warehouse). VAL contains information on all consultations in primary and specialist outpatient care, as well as hospitalizations. Patients visiting the cluster center will be identified through center codes linked to each encounter. VAL also contains issued diagnoses and procedures, which will help us to evaluate eligibility for screening as well as occurrence of new diagnoses of CKD during the trial. VAL also allows identifying accepted referrals to nephrologist care. Finally, other necessary information from VAL includes demographic information (age and sex), migration to/from Stockholm (which allows for censoring participants when their residency changes) and ascribed primary care center (to explore the proportion of patients attending their assigned primary care center or another one).
2.The repository of laboratory data. Three laboratory companies (Unilabs, Synlabs and Karolinska) perform the vast majority (> 95%) of clinical chemistry laboratory tests of the region, including the totality of SLSO centers included in our trial. From this repository, we will extract information on all performed analyses of plasma/serum creatinine, cystatin C and albuminuria (dipstick and urinary albumin to creatinine ratio). For each measurement we will have information on the date and time of measurement, units, method used, reference interval, and the department/unit that requested the test. In this way, we can identify with precision which tests were ordered from the centers in our trial.
3.Linkage with two registries coordinated by the Swedish National Board of Health and Welfare: (a) The Prescribed Drug Registry [21] containing all pharmacy dispensations of prescribed drugs. This will allow for the evaluation of use or initiation of recommended CKD-modifying agents; and (b) The Population Registry [21] with information on vital status and, when applicable, reported cause of death as a censoring event.
4.Via linkage with TakeCare intelligence warehouse, which administers the electronic healthcare records system of the region, we will obtain information on clinical parameters such as height, body weight, blood pressure or smoking habits of patients seeking care in the centres involved in our trial. This will not be necessary to analyse the outcomes of our trial, but it will be used in the next phase where we will explore the impact of CKD recognition and management on health events.
Sample sizeTo estimate the required sample size, we evaluated the processes of CKD care in the 66 primary healthcare centers entering this trial during 2019. This information was available to us from the SCREAM repository. We chose the year prior to the COVID-19 pandemic to have more representative estimations of health care use not affected by lock downs and policies during the pandemic. We believe this information is rather unique and provide it as a supplementary information for use by other researchers in their power considerations for future trials (Supplementary table).
During 2019, 662 995 unique individuals visited the primary healthcare centers included in the trial at least once. The size of the centers varied, as well as the number of patients with an indication for CKD screening in them. In addition, creatinine and albuminuria testing and re-testing rates were different across centers. Some primary care centers were in residential communities/neighborhoods of generally older age, whereas other centers tended to receive patients of younger age, hence with less indications for CKD screening. Of all people visiting the health care centers, 92 011 were eligible for screening/testing of kidney function based on their comorbidity profile: 66% took a creatinine test, and 34% took an albuminuria test. Of people with first detected eGFR < 60 ml/min/1.73 m [2] or albuminuria > 30 mg/L, 59% and 28% were retested for creatinine or albuminuria, respectively, within 3–6 months.
Based on the testing and re-testing rates of participating centers in 2019, the study has 85% power to detect an absolute improvement in our primary study outcome of 13% points with an alpha of 5%, and similar power to detect differences in the first three secondary outcomes. The effect size was estimated by assuming that the intervention would increase the proportion being tested by 20–30% (relative increase). For the primary outcome we used a conservative estimate of 20%, hence from 0.66 to 0.79. We assumed a recruitment period of 12 months and a total of 33 health center (clusters) per arm with the same number of patients as in 2019. We estimated the intraclass correlation for the outcomes to be 0.1 (as quantified in the UK trial by Major et al. [22]).
RandomizationRandomization was restricted to ensure (approximate) balance on two factors that differed across the participating centers: the number of people that visited them in 2019 with an indication for CKD screening, and the proportion of these people that were tested for albuminuria. In total, 62 of the centers had < 3000 patients visiting them in 2019 with an indication for CKD screening. Four centers, however, had a much larger eligible population, with 5434, 4712, 3762, and 3286 patients. We first created 10,000 randomizations of the 62 “non-large” centers, with 31 in each arm. We then created all possible permutations of the four large centers such that two were in one arm and two were in the other arm. There are six possible permutations which achieve this (AABB, ABAB, ABBA, BAAB, BABA, BBAA). These two sets (10,000 randomizations of the 62 large centers, and 6 randomizations of the smaller centers) were combined to give us 60,000 potential randomizations and then we mirrored these allocations to give us 120,000 randomizations (that is, reversed the allocation of intervention and standard arms). For each potential allocation we calculated the average proportion (within each randomization arm) of people tested in 2019. This was calculated on a center level, not a population level, so the denominator was 33 in each arm. If the difference in the average proportion between the arms was greater than 3.7%, we discarded the allocation. The figure of 3.7% was chosen because, under different randomly drawn sets of the 10,000 allocations, 3.7% ensured that approximately, but less than 10% of allocations, were discarded. Furthermore, it was felt that 3.7% was small enough such that if there was indeed a between-arm difference of 3.7% we would still be confident that the two arms were similar enough to be confident of drawing unbiased conclusions form the results. Finally, one allocation was randomly selected from the remaining ~ 108,000 allocations.
The code to perform this randomization was given to an independent statistician, outside of the study team, who changed the random seed that selected the 10,000 allocations, and the random seed that selected the final allocation. When the code was run, 10,348 (8.6%) allocations were removed due to a difference between the arms in 2019 test rates.
Post-randomization change and sample size re-estimationFour days after performing this randomization, the study team was informed that two of the 66 centers had been merged into one administrative unit (although still being two geographically separate sites). Due to this merge, we will not be able to retrieve outcome data at a lower level than the merged center. As by chance these two centers had been assigned to the same arm, we decided to proceed with the trial under the original randomization. We note here that the two original restrictions still held after the merger of these centers: the eligible population in 2019 of the new merged center was 1,455, which means it ranked 24th (of 65) in size; the difference between the two arms in terms of 2019 test rates became 3.4% (< 3.7%). The trial will then analyze 65 centers (33 in the standard arm and 32 in the intervention). Table 2 gives the originally calculated power, and the power that we now have after the administrative merging of two centers, conservatively assuming 32 clusters per arm.
Table 2 Power calculationStatistical analysisAll analyses will be performed on an ITT basis, where each center is analyzed according to the arm it was assigned to at randomization. The primary analyses will be performed at the level of the individual patient. We will use logistic regression, with a random effect to account for the clustering by healthcare center and including a binary variable to indicate randomized treatment allocation. We will additionally include the baseline level of testing in the center (proportion in 2019), and a binary variable indicating the size of the center (large or not-large), as both of these were used in the randomization procedure. This analysis will produce a relative measure of effect, namely an odds ratio. We will also present an absolute measure of the intervention effect.
The primary outcome will also be analyzed at the center level, adjusted for the same variables described above: 2019 testing proportion and center size.
The same approach will be used for the secondary outcomes. To test whether repeated clinic visits lead to changed behaviour, we will perform two subgroup analyses. First, by the number of visits a participant makes during the recruitment phase: 1 visit vs. more than 1 visit. Second, by sex: men vs. women. Exploratory analysis will use a time-to-event outcome for each participant, instead of a simple binary variable calculated at 12 months. No other pre-planned subgroup analyses will be performed, nor are any interim analyses planned. As this study uses routinely collected data we do not anticipate missingness and have no plans to impute missing data. A detailed description of the methods to be used will be given in a Statistical Analysis Plan, which will be uploaded to clinicialtrials.gov prior to the time of last data collection and before any analysis has been performed.
Declarations Ethics approval and consent to participateWe have obtained approval to conduct this trial by the Swedish Ethical Review Authority (EtikPrövningsMyndigheten, EPM, registration number 2023-03537-01). The EPM will be informed of any changes in the study protocol in accordance with applicable requirements. Because the goal of this pragmatic intervention is to improve current clinical practice by ensuring adherence to current recommendations and protect participants from potential risks of CKD underdetection, it was deemed by the Swedish Ethical Review Authority that it was not necessary to obtain informed consent from individual patients potentially accessing those primary care centers during the study. We contacted the head of all 66 primary healthcare centers to inform them of this trial and give them the option of having their center opting out. Patients at sites randomized to standard of care/no extended ALMA will obtain health care through currently available best clinical practice.
1.Data management and data access.
Data will be analyzed and stored at the Department of Medical Epidemiology and Biostatistics at Karolinska Institutet. The trial dataset will not be available for public use as it would breach GDPR regulations of patient’s privacy. Data can be available on reasonable request to investigators for proposals that comply with GDPR, national and institutional regulations for data sharing and collaborative research.
2.Dissemination policy.
We will present results of the study to Region Stockholm and the participating centers and seek to present and publish results of the intervention on the outcomes described here at national/international conferences and in a peer-reviewed scientific journal. Authorship will be decided on intellectual contribution to the research (and not due to any financial incentives), and we have no plans to use professional writers.
3.Steering committee.
Decisions regarding the conduct of the trial, data management and analysis will be considered by the Trial Steering Committee, which consists of Profs. Juan J Carrero, Stefan Jacobsson and Arvid Sjölander, Assoc. Prof Marie Evans, Dr. Jacob Andersson-Emad and Stephen Nash. As all participants will receive standard care under the direction of their regular clinician we do not anticipate adverse events and do not require a Data Monitoring Committee.
4.Funding declaration.
The study is financed by grants from the Swedish state under the agreement between the Swedish government and the county councils, the ALF-agreement (FoUI-986028), and by the Swedish Research Council (2023 − 01807). We also acknowledge donations from AstraZeneca and Boehringer Ingelheim to Danderyd University Hospital. The funders have no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Competing interestsArvid Thunholm is an employee of Blackwell Medtech, the company that developed and manages the ALMA platform. None of the rest of investigators involved in the study have any financial interest or possible benefit from the results of this trial.
5.Author contributions and consent for publication.
Study concept: J.J and S.H.J; Writing of the first draft: J.J.C and J.A.E; First round of revisions: S.N; Second round of revisions and final approval of the Ms: Rest of authors. All authors consent to submit this Ms for publication.
留言 (0)