Gitelman Syndrome is a rare autosomal recessive hereditary tubular disorder characterized by metabolic alkalosis, hypokalemia, hypomagnesemia [1], and normal blood pressure [2], first described in 1966 by Gitelman and his colleagues [3]. GS is caused by loss of function mutations in the SCL12A3 gene, which encodes the thiazide-sensitive sodium chloride cotransporter that reabsorbs sodium chloride in the apical membrane of the distal convoluted tubule [4, 5]. Over 350 variations in the SLC12A3 gene have been identified until now [6]. The majority of these are missense mutations, but splicing, nonsense mutations, and deletions and duplications have also been identified (ref: Vargas-Poussou et al. JASN 2011, 22: 693–703).
Hypokalemia, a key manifestation of GS, is a result of two underlying mechanisms. First, loss of function of the sodium chloride cotransporter results in decreased sodium chloride reabsorptionin the distal convoluted tubule and increased sodium delivery to the downstreamconnecting tubule and cortical collecting duct, which comprise the aldosterone-sensitive distal nephron. Increased sodium reabsorption by the epithelial sodium channel provides an increased driving force for potassium secretion through the potassium secretory channels, ROMK (Renal Outer Medullary Potassium) and BK (Big Potassium, also known as Maxi-K). This is potentiated by volume depletion, which activates the epithelial sodium channel via aldosterone and angiotensin II, as well as increased tubular flow, which stimulates the epithelial sodium channel and BK [4].Second, hypomagnesemia relieves gating of ROMK by magnesium, allowing increased potassium efflux from the principal cells of the aldosterone-sensitive distal nephron [7].
Although hypomagnesemia is another key manifestation of GS, its underlying cause is not very well understood. The TRPM6 (transient receptor potential cation channel subfamily M member 6) magnesium channel, which is located on the apical membrane of distal convoluted tubule epithelial cells, reabsorbs magnesium in that segment (ref: de BaaijPhysiol Rev 2015, 95: 1–46). In mice that are chronically treated with thiazide diuretics, which inhibit the sodium chloride cotransporter, or which have genetic deletion of the sodium chloride cotransporter, there is downregulation of TRPM6, possibly due to the atrophy of the distal convoluted tubule that occurs with loss or inhibition of the sodium chloride cotransporter (ref: Nijenjuis T et al., J Clin Invest 2005, 115: 1651–1658). Decreased TRPM6 in the distal convoluted tubule could explain renal magnesium wasting and hypomagnesemia.
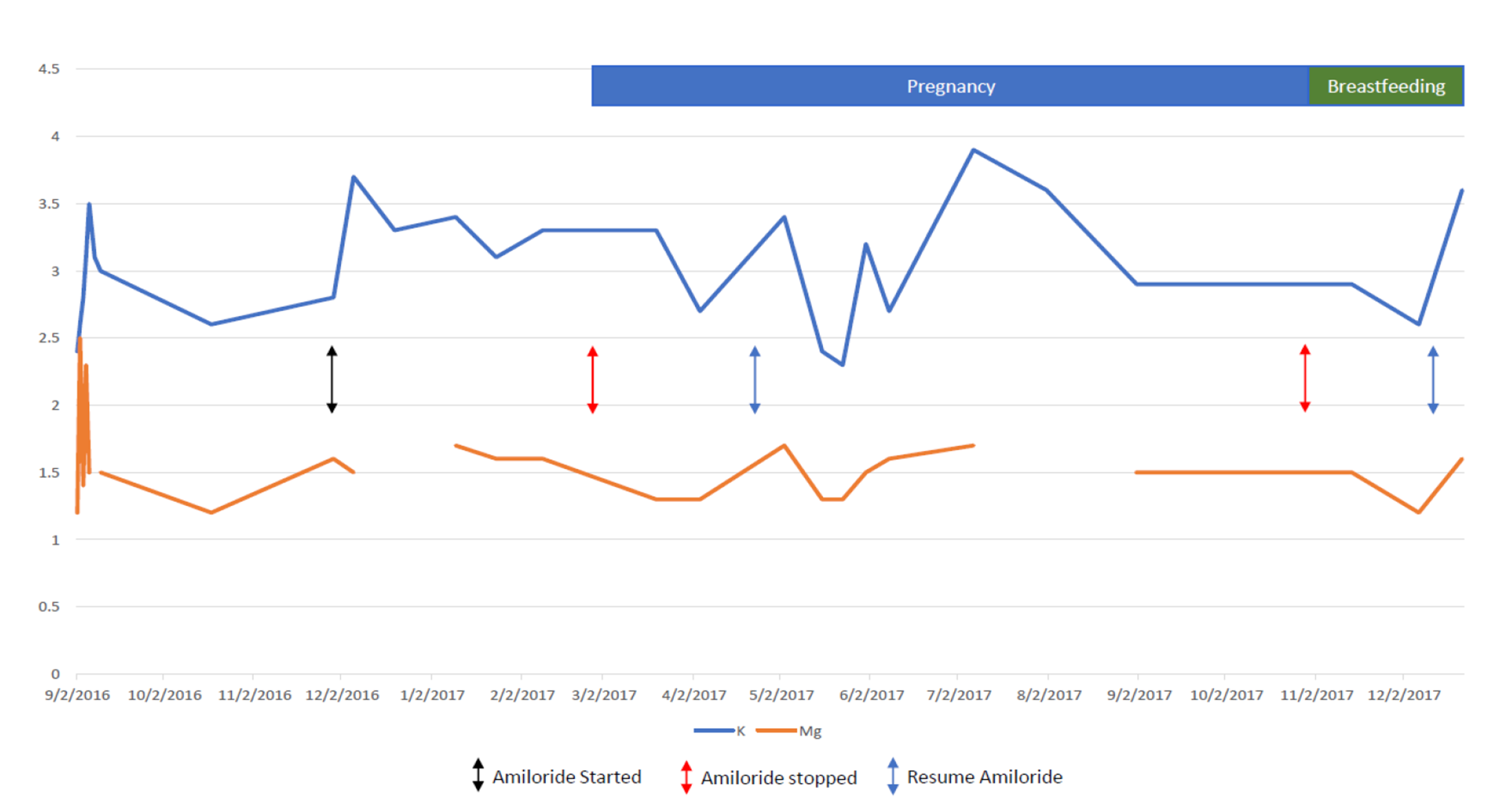
Potassium and magnesium supplementation is the basis of GS treatment. However, in cases where electrolyte level correction via supplementation is insufficient, potassium-sparing diuretics such as amiloride, spironolactone, and eplerenonemay be useful [8]. Amiloride is a potassium and magnesium-sparing diuretic that acts primarily by blocking sodium transport in the aldosterone-sensitive distal nephron, which in turn inhibits sodium-potassium exchange [9]. Physiologically, sodium reabsorption in the distal tubules via epithelial sodium channels (ENaC) results in apical membrane depolarization, which acts as a driving force for potassium secretion [7]. Therefore, asamiloride directly inhibits ENaC, it hyperpolarizes the apical membrane which decreases the electrochemical driving force for potassium efflux, and results in its antikaliuretic potassium-sparing effects [10]. It is classified as a category B drug by the Food and Drug Administration in terms of teratogenicity. Although teratogenicity evaluation in rodent models is reassuring, amiloride is concentrated in breast milk and thus it is not recommended to be used during breastfeeding unless it is strongly indicated [11]. To the best of our knowledge, there are no reports of amiloride levels in human breast milk, and assays for measuring amiloride concentration in breast milk are not commercially available. However, in our case, the infant’s relatively elevated serum potassium, and low urinary potassium and magnesium concentrations, suggested a systemic effect of amiloride.
Pregnancy is associated with many renal physiological changes, including positive balance of both sodium and potassium, which could be complicated by renal sodium and potassium losses in Gitelman’s syndrome. Renin, angiotensin II, and aldosterone levels are known to increase during pregnancy. However, the increased progesterone levels during pregnancy would counteract the effects of increased activation of the renin angiotensin-aldosterone system on potassium excretion due to its anti-mineralocorticoid effects [12]. Nonetheless, the compensatory mechanism of progesterone on potassium excretion may be inadequate to prevent excessive potassium excretion in pregnant women with GS [12]. Moreover, potassium wasting may be exacerbated during pregnancy due to additional physiological changes, such as increased fetal demand, and vomiting [13].
Although in rodent models, magnesium deficiency during pregnancy has been associated with significantly increased neonatal mortality and morbidity [14], it appears that GS represents no significant risk to the fetus in human clinical reports [15]. Thus, the main therapeutic goal for patients with GS is to maintain their potassium and magnesium serum levels and prevent symptoms [13, 15]. Additionally, electrolyte correction is critical to prevent potentially fatal maternal complications such as ventricular arrhythmias.
Although the cornerstone for GS treatment during pregnancy is liberal salt intake, potassium and magnesium supplementation, it appears that it is more difficult to achieve stable normal serum levels of these electrolytes in pregnant patients [16].
The use of medications such as Spironolactone, amiloride, and eplerenone has been rarely reported due to their controversial safety profile during pregnancy [16]. In the rat, the epithelial sodium channel is activated during pregnancy to allow net positive sodium balance and the plasma volume expansion that is characteristic of pregnancy, and pharmacological inhibition of the epithelial sodium channel prevents plasma volume expansion (West C et al. Am J PhysiolRegulIntegr Comp Physiol 299: R1326-R1332, 2010). However, amiloride has been successfully used for managing hypokalemia in a few pregnant patients with Bartter’s Syndrome [17] and Liddle’s Syndrome [18].
We are aware of only four previous reports of amiloride use during pregnancy in GS patients [19,20,21,22]. In all cases amiloride was used due to persistent hypokalemia unresponsive to potassium and magnesium supplementation alone. All pregnancies resulted in healthy newborns; however, one pregnancy required labor induction at 38 weeks of gestation due to oligohydramnios [19]. The use of amiloride in our case was necessary to maintain adequate potassium and magnesium serum levels in our patient and thus to prevent further serious complications.
Although amiloride is classified by the FDA as category B in terms of teratogenicity, no randomized clinical trials have been conducted to investigate their actual safety profile in humans during pregnancy and breastfeeding.
In conclusion, we report a safe and effective use of amiloride during pregnancy in a GS patient which facilitated control of her persistent hypokalemia and hypomagnesemia despite oral supplementation therapy. However, due to the uncertainty regarding its safety during breastfeeding and the limited available literature, we cannot make a definitive recommendation for amiloride use during this period. Accordingly, we suggest that more clinical studies are needed to evaluate the safety profile of amiloride therapy during pregnancy and breastfeeding.








留言 (0)