Materials
N-Methyl-4-Phenylpyridinium Iodide (MPP+), Trimethylamine N-Oxide (TMAO), Tunicaymycin, Sodium 4-Phenylbutyrate (4-PBA) were obtained from Aladdin Inc (Shanghai, China), 1-Methyl-4-phenyl-1, 2, 3, 6- tetrahydropyridine (MPTP) and MK-2206 2HCL were purchased from MACKLIN (Shanghai, China). Dulbecco's modified Eagle's medium (DMEM)/Nutrient Mixture F-12 (DMEM/F12), 0.25% trypsin, penicillin–streptomycin and fetal bovine serum (FBS) were purchased from Gibco (Grand Island, NY). The primary antibody against CHOP, AKT, p-AKT (Ser473), p-GSK-3β (Ser9) was purchased from Cell Signaling Technology (MA, USA). Anti-GSK-3β antibody was from Abcam (Cambridge, MA, USA). The primary antibody against β-catenin, JNK, p-JNK, Caspase-3/Cleaved caspase-3, Bax, Bcl-2, Tyrosine Hydroxylase (TH) were from WanleiBio (Shenyang, China). Hoechst 33,342, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazoliumbromide (MTT), the kits of5,5′,6,6-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanineiodide (JC-1), Pi-dium (PI), and DCFH-DA, N-Acetyl-L-cysteine (NAC) and the primary antibody against IRE-1α, p-IRE-1α, eIF-2α, p-eIF-2α, GRP78 were from Beyotime Biotechnology (Shanghai, China). The primary antibodies against β-actin and all of the secondary antibodies were obtained from ProteinTech Inc (Wuhan, China). The reagents used in this research were all commercially available and meet biochemical quality standards.
Periplaneta americana L. extract preparation
The dry body of Periplaneta americana L. (P. americana) was from GoodDoctor Pharmaceutical Group (Sichuan province, China), identified by characteristics and confirmed to be genuine by thin-layer chromatography (TLC). Specifically, it is reddish brown in color and has wings longer than the end of its abdomen and a large butterfly shaped brown stripe in the middle of the pronotum from appearance. The sample and the reference substance display the same spots at the corresponding positions when identified by TLC. To prepare the extract for experimental use, P. americana was ground to powder and degreased in petroleum ether for 24 h. After removing the petroleum ether completely, added 75% ethanol to the degreased powder until it swelled, and then transferred it to a percolation tube to percolate extraction within five-fold times of 75% ethanol for 48 h. The flow rate was controlled at 3 mL/min. The ethanol extract was collected and centrifuged at 12,000 rmp for 10 min, and the supernatant was filtrated through 0.45 μm microporous membrane. The filtered extract was then evaporated under reduced pressure followed by lyophilization to produce dried extract with yield of 15.7%.
LC–MS/MS conditions
The Agilent ultra-high performance liquid chromatography 1290 UPLC system with ACQUITY UPLC® HSS T3 (2.1 × 100 mm, 1.8 µm) chromatographic column (Waters, Milford, MA, USA) was used to perform the LC–MS/MS analyses. The flow rate was controlled at 0.3 mL/min, the temperature was set at 40 ◦C and the injection volume was 2 μL. The mobile phase consisted of 0.1% formic acid in acetonitrile (B1) and 0.1% formic acid in water (A1) under positive ions mode. The multi-step linear elution gradient program was: 0–1 min, 8% B1; 1–8 min, 8%-98% B1; 8–10 min, 98% B1; 10–10.1 min, 98%–8% B1; 10.1–12 min, 8% B1. Under negative ions mode, the mobile phase is acetonitrile (B2) and 5 mM ammonium formate in water (A2). The multi-step linear elution gradient program was: 0–1 min, 8% B2; 1–8 min, 8%-98% B2; 8–10 min, 98% B2; 10–10.1 min, 98% -8% B2; 10.1–12 min, 8% B2.
A Thermal Q Exactive Focus mass spectrometer together with the Xcalibur software (version 4.1) was used to get MS and MS/MS data. During each acquisition cycle, the mass range was from 100 to 1500, and the top three for each cycle were screened, with corresponding MS/MS data acquisition. Spray Voltage: 3.5 kV (positive) or -2.5 kV (negative). Sheath gas flow rate: 40 arb, Aux gas flow rate: 10 arb, Capillary temperature: 325 °C, Full scan resolution: 70,000. HCD was used for secondary fragmentation with a collision energy of 30 eV, MS/MS resolution: 17,500.
Cell culture
The SH-SY5Y cell line used in our study was from American Type Culture Collection (ATCC, USA). The cells were cultured in flasks containing DMEM/F12 modified with 10% FBS and 1% penicillin/streptomycin, and maintained in an incubator at 37 °C in 95% air/5% carbon dioxide. The cells were passaged at 48 h per interval and seeded into 96-well plates, 24-well plates or 6-well plates according to the experiment design.
Cell viability analysis
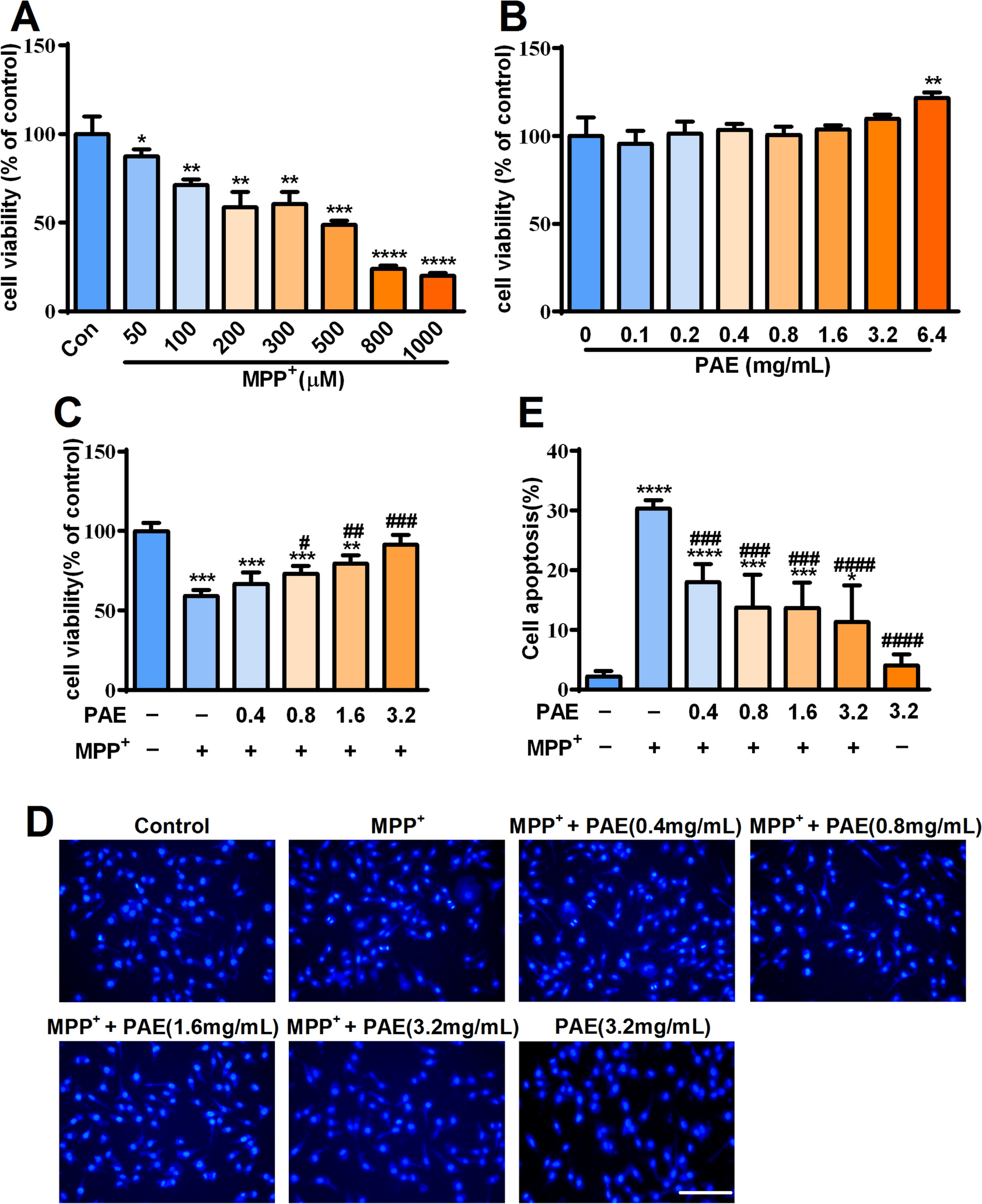
Cell viability was determined by the MTT assay. The SH-SY5Y cells were cultured in 96-well plates at a density of 5 × 103 cells/well for 24 h. Then, the culture medium was discarded and new medium containing different concentrations of MPP+ or PAE was added for another 48 h. To study the effect of PAE on MPP+ toxicity, the cells were pretreated with various concentration of PAE for 2 h, followed by exposure to MPP+ for another 48 h. In the latter experiments, for the cells needed to be treated with TMAO or tunicaymycin, they were added to the wells simultaneously with PAE. For the cells needed to be treated with NAC or 4-PBA, added these compounds into the well for 2 h and then exposed the cells to MPP+ (500 μM) for another 48 h. Subsequently, the DMEM medium containing MTT (0.5 mg/mL) was added to each well and then incubated at 37 °C for 4 h. The medium was then replaced by 200 μL dimethyl sulfoxide (DMSO) to dissolve the formazan product. The optical density (OD) was measured at 570 nm with a multi-functional microplate reader (Synergy HT, Bio-TEK instruments Inc., USA). The cell viability was expressed as a percentage of the absorbance of untreated cultures.
Hoechst 33,258 staining
The cells were cultured in 48-well plates at a density of 2 × 104 cells/well. They were pretreated with different concentrations of PAE for 2 h and then incubated for another 48 h in the presence or absence of MPP+ (500 μM). For the cells needed to be treated with TMAO, tunicaymycin, NAC or 4-PBA, added them to the well in the way as described above. The cells were then fixed with 4% paraformaldehyde for 20 min at room temperature. After being washed by PBS, the cells were stained with Hoechst 33,258 (10 μg/mL) for 15 min, and the nuclear changes were observed with an inverted fluorescence microscope (Nikon, Japan) [27].
Calcein AM-PI staining
The Calcein AM-PI Staining Kit (Beyotime Biotech, Shanghai, China) was used to detect live and dead cells [27]. Briefly, SH-SY5Y cells were pretreated with PAE for 2 h and incubated with or without MPP+ (500 μM) for another 48 h. For the cells needed to be treated with TMAO, tunicaymycin, NAC or 4-PBA, added them to the well in the way as described above. After the treatment, cells were then stained with Calcein AM-PI solution for 20 min at room temperature. After being washed with PBS, the fluorescence was detected with an inverted fluorescence microscope.
Flow cytometric analysis
Flow cytometric analysis with Annexin V-FITC/PI double staining was performed to assess the cell apoptosis [28]. Briefly, SH-SY5Y cells were seeded in 6-well plates and treated as described above. Subsequently, the cells were digested and centrifuged at 1000 rpm for 5 min. Afterwards, the cells were suspended with 500 μL binding buffer and incubated in dark with Annexin V-FITC and PI for 15 min at room temperature. Cell fluorescence was analyzed by a flow cytometer (BD FACSVerse™ flow cytometer, BD Biosciences, San Jose, CA, USA). Viable cells, early apoptotic cells, late apoptotic cells and cellular debris were identified as Annexin V−/PI−, Annexin V+/PI−, Annexin V+/PI+ and Annexin V−/PI+, respectively.
Intracellular ROS measurement
Cells were cultured in 24-well plates at the density of 2 × 104 cells per well. The treatment schedule was same as described above. For the cells needed to be treated with MK-2206, 5 μM MK-2206 2HCL was added to each well simultaneously with PAE. After the treatment, the cells were incubated with DCFH-DA (10 μM) for 20 min at 37 °C in dark [29]. Subsequently, cells were washed with PBS and the fluorescence of DCFH-DA was imaged under a fluorescence microscope (Nikon, Tokyo, Japan).
Mitochondrial membrane potential measurement
The mitochondrial membrane potential (Δψm) was measured by JC-1 kit (Beyotime Biotech) [30]. JC-1 is a cell-permeant dye that can accumulate within mitochondria. It is widely used to detect Δψm. In normal cells, JC-1 aggregates in the mitochondrial matrix to form J-aggregates and produce red fluorescence. However, in the apoptotic cells, Δψm is decreased and JC-1 cannot be gathered in the mitochondrial matrix, thus JC-1 exists in the form of a monomer and produces green fluorescence [31]. The ratio of red/green fluorescence can be used to reflect the change of Δψm. As described above, the cells were pretreated with various concentration of PAE for 2 h and then exposed to MPP+ for another 48 h. For the cells needed to be treated with MK-2206, it was added to each well simultaneously with PAE at a final concentration of 5 μM. Afterwards, the cells were washed and incubated with JC-1 staining solution for 20 min at 37 °C in dark. Finally, the cells were washed with JC-1 staining buffer three times and the fluorescence was imaged with an inverted fluorescence microscope (Nikon, Tokyo, Japan). For red fluorescence, the excitation and emission light was 525 nm and 590 nm. For green fluorescence, the excitation and emission light was 490 nm and 530 nm, respectively.
Immunofluorescence staining
Cells were fixed with 4% paraformaldehyde for half an hour followed by PBS washing, and then blocked with 10% goat serum containing 0.3% Triton X-100 for half an hour at room temperature [29]. Afterwards, the cells were incubated with the rabbit anti-p-IRE1α antibody (1:200), anti-p-eIF2α antibody (1:200), anti-GRP78 antibody (1:200) and anti-CHOP antibody (1:200) at 4 °C overnight. On the next day, the cells were washed by PBS for three times and then incubated with Cy3-labeled goat anti-rabbit IgG (1:100) at room temperature in the dark for 1 h. After removing the secondary antibody and washing with PBS, the cells were stained with DAPI for 15 min. Cell fluorescence was imaged and captured using a confocal microscope. Six views of each group were selected randomly and used for the following analysis.
For the immunostaining of brain tissues in the latter experiment, the procedure was similar to that of cultured cells. In brief, the mouse brain was removed rapidly after perfusion and post-fixed overnight in 4% paraformaldehyde at 4 °C. Subsequently, the tissue was dehydrated sequentially with 20% and 30% sucrose solution until it sank to the container bottom. The coronal sections (15 μm) containing striatum and substantia nigra were obtained using a freezing microtome (Leica). After being washed with PBS for three times, the cryosections were blocked with 10% goat serum containing 0.3% Triton X-100 at room temperature for 1 h. Then sections were incubated with primary antibodies overnight in 10% normal goat serum at 4 °C. Subsequently, the sections were rinsed thress times with PBS and then incubated with the fluorescence-labeled secondary antibody in dark for 1 h at room temperature. The sections were washed again and stained with DAPI for 15 min. Finally, the sections were coverslipped with 50% glycerine and photographed under a microscope.
Animal experiments
Male C57BL/6 mice (8 weeks old, specific pathogen free) were obtanied from Slike Jingda Laboratory Animal Co., Ltd (Hunan, China). The mice were housed under standard laboratory conditions (temperature 22–26 °C, humidity 50–70%, 12 h light/dark cycle) with water and food provided ad libitum. Before the experiments, they were allowed to acclimate to laboratory environment for one week. All experiments were conducted in accordance with the guidance for the Care and Use of Laboratory Animals published by the US National Institutes of Health [32] and the protocols approved by Animal Care and Use Committee of the Southwest University.
Mice were randomly divided into four groups: the vehicle group, the MPTP group, the MPTP + PAE group, the MPTP + PAE + MK2206 group. Mice in the vehicle group and MPTP group were administered with saline intragastrically (i.g.), and the other two groups were given the same volume of PAE (120 mg/kg, dissolved in saline, i.g.) from day 1 to day 14. From the third day to the seventh day, two hours after the administration of saline or PAE, the mice in the vehicle group were given saline intraperitoneally (i.p.) (10 mL/kg) and mice in the other three groups were given the same volume of MPTP (30 mg/kg, i.p.). For mice in the MPTP + PAE + MK2206 group, the Akt inhibitor MK-2206 2HCl (100 mg/kg, i.g.) was administrated every other day starting from day 3, for a total of six times. Behavioral tests were carried out on day 15, and then the mice were sacrificed by overdosing 5% isoflurane and the tissues were collected for the following experiments.
Rotarod test
The rotarod test is commonly used to assess the motor coordination and balance in rodents [29]. Mice were placed on a rotarod rod (Ji-Nan Yiyan company) and the rolling speed was adjusted so that it gradually increased to 30 rpm within 30 s. The latency of each mouse to fall off the rotarod was recorded. The maximum cutoff time was 180 s. If the animal had not fallen after 180 s, the latency would also be recorded as 180 s. Each mouse was tested for three times with an interval time of at least half an hour. To reach optimal performances, the mice were subjected to three consecutive days of adaptive training before the formal experiment.
Pole test
The pole test was commonly used to detect the coordination ability of mice [33]. A straight wooden rod with the diameter of 0.8 cm and height of 50 cm was used in the pole test. A small wooden ball wrapped with bandage gauze was placed at the top of the rod to prevent the mouse from slipping [29]. Mice were trained in three test trials for two days prior to the formal experiment. In the formal test, mouse was placed on top of the straight rod, and the time it took to completely turn around and face the floor was recorded as T-turn (time to turn) and the total time required to arrive at the bottom of rod was recorded as T-down (time to climb down). Each mouse was tested three times with an interval of at least half an hour.
Western blot analysis
Western blot analysis was carried out as previously described [34]. In brief, the harvested cells or brain tissues (striatum and substantia nigra) were lysed with RIPA buffer containing the proteinase and phosphatase inhibitors. The samples were subjected to an ultrasonic cell disruptor in order to be homogenized completely, and then centrifuged at 12,000 rpm for 15 min at 4 °C. The supernatant was collected and the protein concentration was quantified with BCA Protein Assay Kit (Beyotime Biotech). Equal amounts of protein (30 μg) were separated by SDS-PAGE gel and then electrotransferred onto PVDF membranes (Invitrogen). The PVDF membranes were blocked in 5% fat-free milk at room temperature for 2 h, then the bands were cut according to their molecular weight and incubated overnight with primary antibodies at 4 °C. Subsequently, the membranes were washed three times with TBST for 10 min each time, and then probed with the horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG secondary antibodies for 1.5 h at room temperature. Finally, the bands were visualized using an enhanced chemiluminescence (ECL) system (Perkin Elmer). The results were expressed the ratio relative to the band intensity of control group.
Statistical analysis
Results were presented as means ± SD. Statistical comparisons were performed with GraphPad Prism 7 (San Diego, CA, USA) using one-way analysis of variance (ANOVA) followed by Tukey’s test for post hoc comparisons. In all cases, p < 0.05 was considered as statistically significant.









留言 (0)