
記住我

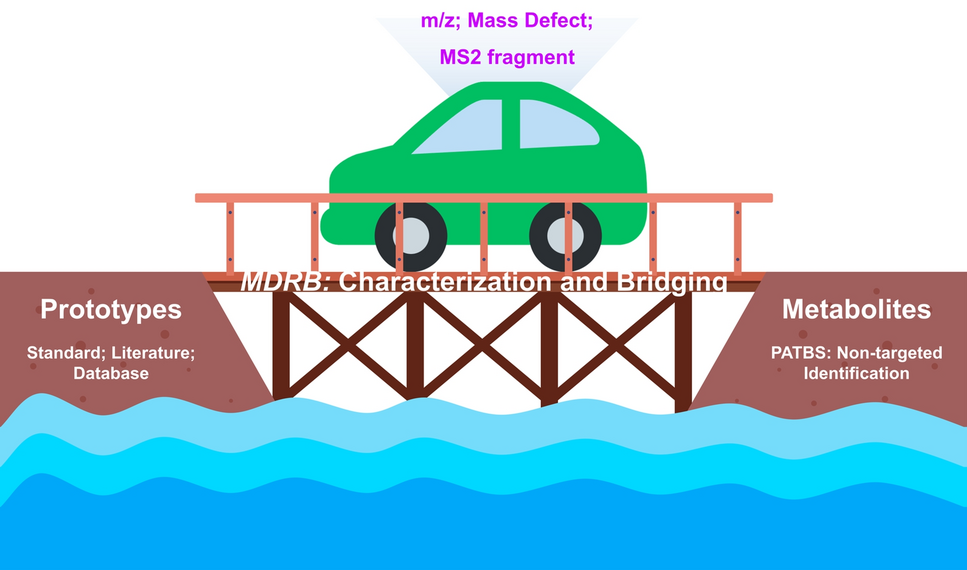







In order to realize the transformation attribution of relevant components in TCM and to comprehensively construct important in vivo metabolic profiles, we established a novel strategy for the identification, characterization, and bridging analysis of in vivo metabolites (Fig. 1). The first step is to establish two lists, including prototype list and metabolite list. Identification of prototypical components was carried out by the fingerprint analysis of CKXR to generate a prototype list. The metabolite list mainly relies on the PATBS non-targeting technique. This technique is mainly based on retention time (RT) and m/z, as well as setting a narrow mass window (10 ppm) and time window (± 0.5 min), comparing the blank and sample spectra, and realizing the background subtraction of the biological samples according to a certain subtraction ratio. In this case, the mapping and maximize the obtainment of CKXR related components in mice can be simplified, in order to constitute a metabolite list. Secondly, we utilized the MDRB strategy to achieve progressive correlation of in vivo and in vitro components, which is mainly divided into three steps: prototypes matching, predictable metabolites matching, and unpredictable metabolites matching. This matching is mainly based on MS1, MS2, RT, m/z, and mass defect information, which is assigned by different comparison and screening methods to construct the association between prototypes and metabolites. Employing this holistic analysis strategy, we can identify and characterize the metabolites and map the metabolic networks of TCM in vivo, offering a visual representation of its overall mechanism of action.
Fig. 1
A workflow of using UPLC–HRMS, PATBS and the mass dataset relevance bridging (MDRB) technique for the discovery and identification of metabolites
Recognition of prototypic components and metabolites in vivoThe CKXR sample was detected to obtain the high-resolution total ion chromatogram (TIC) in positive and negative ion modes. Different components exhibited different retention behaviors on the column and produce characteristic MS2 fragmentation patterns. The identification and characterization of the components of CKXR were carried out based on various methods, such as the identification of the reference substances, and the comparison of the literature and databases [Pubchem (https://pubchem.ncbi.nlm.nih.gov/) and Chemspider databases (https://www.chemspider.com/Default.aspx)]. We found a total of 199 components in CKXR (Fig. 2A, B). Among them, 96 substances were detected in the positive ion mode, while 136 substances were detected in the negative ion mode, and 33 substances responded to both positive and negative ion modes. Detailed MS information of the relevant components of CKXR is shown in Table S2. Our experiment conjectured the possible structures of 128 components, of which 15 (including amygdalin, prunasin, and chlorogenic acid) were characterized by the reference substances or literature. During the process of identification and characterization, we found that CKXR had the highest relative contents of amygdalin and prunasin. Their RT and MS2 information were consistent with the reference substances, and the specific MS2 information was resolved in Figure S1. Finally, we constructed a list of prototypes of CKXR.
Fig. 2
High-resolution total ion chromatogram of CKXR in positive ion mode (A) and in negative ion mode (B). C Full-scan MS spectrum of M254 from PATBS-processed UPLC-HRMS dataset in the negative mode. D–I After PATBS-processed chromatography of full-scan MS analysis of plasma, urine and lung after administration in positive and negative ion mode
Furthermore, we used the PATBS technique for non-targeted selection of peaks of CKXR-related components in biological samples, which can simplify MS information and accurately and completely remove the signals of endogenous interfering substances in full MS datasets [14, 15]. In Fig. 2C, before PATBS treatment, M254 showed low abundance because of the presence of high abundance endogenous components. This might be easily overlooked during MS1 data retrieval. However, MS1 information of M254 was presented in a more effective manner after PATBS technique treatment. PATBS enables comprehensive, non-targeted identification of differential components in samples before and after drug administration. The interference of endogenous components can be effectively excluded, thus rapidly obtaining CKXR-related components in the administered biological samples. We obtained 351 CKXR-related exogenous components, including 41 prototypes and 310 metabolites in mice. 87 CKXR-related components were retrieved from plasma; 53 CKXR-related components were retrieved from lung tissues; 342 CKXR-related components were retrieved from urine; and 42 CKXR-related components were retrieved from feces. All identified CKXR-related components were compiled into a metabolite list. Detailed MS information of CKXR-related metabolites in mice is shown in Table S3 and Fig. 2D–I.
Characterization and bridging between in vitro and in vivo components based on MDRBCharacterization of metabolites usually relies on predictable metabolic pathways, achieved through primary mass number changes and MS2 similarity. However, this process does not cover unpredictable metabolites and often results in the loss of information related to potential active ingredients [29]. Since the mass defect between metabolites and prototypes tends to be within a certain range, mass defect can play a key role in the search for characterization of unpredictable metabolites [30]. In order to efficiently and comprehensively obtain the network of relationships between prototypes and metabolites, we established the MDRB strategy, which synthesizes the predictable changes in the m/z, the mass defect of MS1 and MS2, and the similarity of MS2 fragments for the characterization of metabolites and their correlation bridging with the prototypes. In summary, the characterization and association of the list of prototypes (Table S2) and the list of related components in mice (Table S3) were achieved through a step-by-step matching process. During the matching process, we established an assignment mechanism to facilitate researchers to visualize the intensity of the association between the two (Fig. 3), and a screenshot of the relevant key codes can be found in Figure S2.
Fig. 3
Algorithm assignment process and scoring mechanism
The first step is the characterization of prototypes and isomers of TCM. We set screening standards of RT of ± 25 s and m/z of ± 5 ppm to characterize the prototypes of TCM in mice. The screening standard for isomerism was then m/z within ± 5 ppm error, but RT was not within ± 25 s.
The next step is the characterization of predictable metabolites, which is the products of established and anticipated metabolic pathways. We based the known predictable metabolic responses provided by Metworks 1.3.0.200. Combined with the characteristics of TCM components, we added the deglycosylation reactions that TCM components are easy to undergo within living organisms (Table S4). With the help of databases to predictable metabolites, a catalog of high-resolution m/z values of expected metabolites that undergo one- or two-step metabolic reactions is established. When the m/z of a component in the metabolite list in mice was consistent with the m/z of a prototypes after a predictable metabolic reaction (within ± 5 ppm), it was initially included in the category of possible metabolites of the prototypes and assigned a score of 70, which was further validated. MS2 fragments can provide more information for the association between metabolites and prototypes, and we assign a score to the similarity match hierarchy of MS2 information. When the degree of match is 1, 20 points will be assigned. When the degree of match is greater than or equal to 2, 30 points will be assigned. When the degree of match is 0, 0 points will be assigned. And it is initially concluded that there is no metabolic transformation relationship between the two under consideration.
The final step is the characterization of unpredictable metabolites, which is the products undergo unpredictable metabolic pathways. Here we introduced mass defect to filter of large-scale datasets. Different mass defect ranges (± 10 mmu, ± 25 mmu, ± 50 mmu, and ± 75 mmu) were selected for optimization in order to achieve a more comprehensive and efficient pathway attribution for the prediction of metabolite ground transformation in TCM (Figure S3). The results show that a narrower range, with a reduced proportion of metabolic pathways that can be included (Figure S3A), is able to largely exclude the interference of endogenous components, but the positivity rate is affected to some extent (Figure S3B). The wider range allows for higher positivity rates, but there is often interference that cannot be eliminated when performing multi-component metabolic transformation correlations, leading to increased false positivity rates (Figure S3C). Therefore, we chose ± 25 mmu as MS1 and MS2 mass defect range for unpredictable metabolic pathway matching between prototypes and metabolites. This mass defect range can exploit more effective metabolite information, provide more accurate metabolic transformation relationship, greatly reduce false positive results, and have better practical application value. MS1 Mass Defect Screening Standard: When MS1 m/z of a component in the metabolite list in mice falls within ± 100 amu of MS1 m/z of a prototypes and has a mass defect of ± 25 mmu or less, it is initially included in the range of unpredictable metabolites of prototypes and assigned a score of 50. Matching of MS2 fragmentation information was then performed, with matches and scores assigned consistent with predictable metabolic pathways. MS2 Mass Defect Screening Standard: MS2 fragments of a component in the metabolite list are matched one-to-one with MS2 fragments of the prototypes. When MS2 m/z of the metabolite falls within ± 50 amu of MS2 m/z of the prototypes and the mass defect is within ± 25 mmu, the matching degree will be increased by one accordingly, and 5 points will be assigned for every instance of successful match. Finally, after manually checking for matches of unpredictable metabolites, we set a retention score of 50. In addition, we scrutinized the known metabolic pathways, adding to define that the change in the number of N between prototypes and metabolites was not greater than 3.
Components that do not fall within the scope of the previously mentioned steps are considered elusive in terms of their metabolic pathways. To date, these metabolites have evaded identification of their biochemical processes. It is worth noting that the additive form of components is not limited to the addition or subtraction of hydrogen ions, and we have fully considered the effects of different additive forms (e.g., [M + Na]+, [M + NH4]+, [M + HCOO]−) during the actual matching process, which can be adjusted according to the actual results. Meanwhile, predictable metabolic pathways can equally be adapted to one's own samples. In addition, to increase the confidence of MS2 fragmentation information, we also defined the mass defect between MS1 and MS2, assigning a score of 2 when it falls within ± 50 mmu.
In summary, the main factors of scoring are m/z of MS1, m/z of MS2 and mass defect. Here, we believe that scoring is extremely related to the importance of factors. Factors more valuable to the matching process were assigned higher scores. Specifically, MS1 plays an important role in predictable metabolic pathway. Only when MS1 of the signal to be matched is within the range of conditions can it be considered as a predictable metabolite. At the same time, mass defect is of vital importance for the unpredictable metabolic pathway. Substances with mass defect beyond the allowed range will be regarded as uncommon metabolites and thus be removed from the list of metabolite candidates. MS2 mainly supports the matching process. If MS2 of a signal is similar to that of prototype, the matching would be more reliable. Take metabolite A, and its matching signal (signal B and signal C) as an example. After step 2 process (predictable metabolic pathway matching), signal B and C both could be regarded as predictable prototypes of metabolite A. However, the difference is that signal B could match three MS2 with prototype A, while signal C could only match one MS2 with metabolite A. In this case, prototype B is assigned with 100 scores and prototype C is only assigned with 90 scores. Thus, signal B is more likely to be the predictable prototype of metabolite A.
To more intuitively illustrate the data processing effects of MDRB, we compared the number of metabolites characterized with and without MDRB. A total of 110 metabolites could be characterized manually through matching predictable metabolic pathways, calculation of m/z differences, and comparison of MS2. The rate of manual characterization was 31%. However, utilizing MDRB for data processing analysis, 227 metabolites could be characterized through prototype matching, predictable metabolic pathway and unpredictable metabolic pathway matching, with the characterization rate of 65% (Figure S4A). It can be seen that the use of MDRB can greatly improve amounts and characterization rate of metabolite compounds. Furthermore, we took amygdalin and prunasin as examples, comparing metabolic profiles constructed separately based on manual characterization (Figure S4B) and based on MDRB (Figure S4C). We found that the metabolic profiles were more comprehensive and comprehensive after MDRB treatment. In summary, MDRB tool analysis can increase the number of metabolites characterized and provide more information for constructing metabolic profiles.
The correlation of 227 CKXR-related components in mice was accomplished using the MDRB technique described above. We characterized 66 relevant metabolites in plasma, 222 relevant metabolites in urine, 30 relevant metabolites in feces, and 40 relevant metabolites in lung tissue. Specific matching information is presented in the Table S5. The MDRB technique proposed in this study enables a more comprehensive metabolite characterization and a complete construction of the relationship network of TCM components in vivo and in vitro. The MDRB has a 65% identification efficiency. Of note, by defining the MS2 mass defect, we can achieve more metabolite-prototypes association construction, which is beneficial to adequately characterize the relevant metabolites in TCMs in vivo.
Metabolic transformation relationship of cyanogenic glycosidesFollowing the outlined analytical approach, we procured the scoring and correspondence data (Table S4) pertaining to the constituents linked with CKXR within murine models. Consequently, we were able to delineate a predictive trajectory for metabolic conversions, specifically attributing these processes to the related metabolites of CKXR in mice. By leveraging AUC pooled method, we streamline intricate pharmacokinetic models through consistent criteria, significantly diminishing the burden of sample collection and the investment of time in analysis. This enables us to swiftly and effortlessly uncover the pronounced exposure levels of herbal components within biological systems. Such an efficient approach to pharmacokinetic assessment accelerates research progress while optimizing the monitoring of in vivo behavior of active compounds, making it both cost-effective and resource-efficient [27, 28]. We are concerned that cyanogenic glycosides represented by amygdalin (H88/M197) and prunasin (H97/M215) are the main exposed components of CKXR in mice. Therefore, we analyzed the metabolic characteristics of cyanogenic glycosides, centering on amygdalin and prunasin, and constructed the exposure profile of cyanogenic glycosides in mice (Fig. 4A). Meanwhile, we mapped the ADME metabolic profile of the cyanogenic glycosides of CKXR in mice based on the detection of cyanogenic glycosides in plasma, lung, urine and feces (Fig. 4B).
Fig. 4
Exposed mass spectrum (A) and ADME metabolism profile (B) of cyanogenic glycosides from CKXR in mice
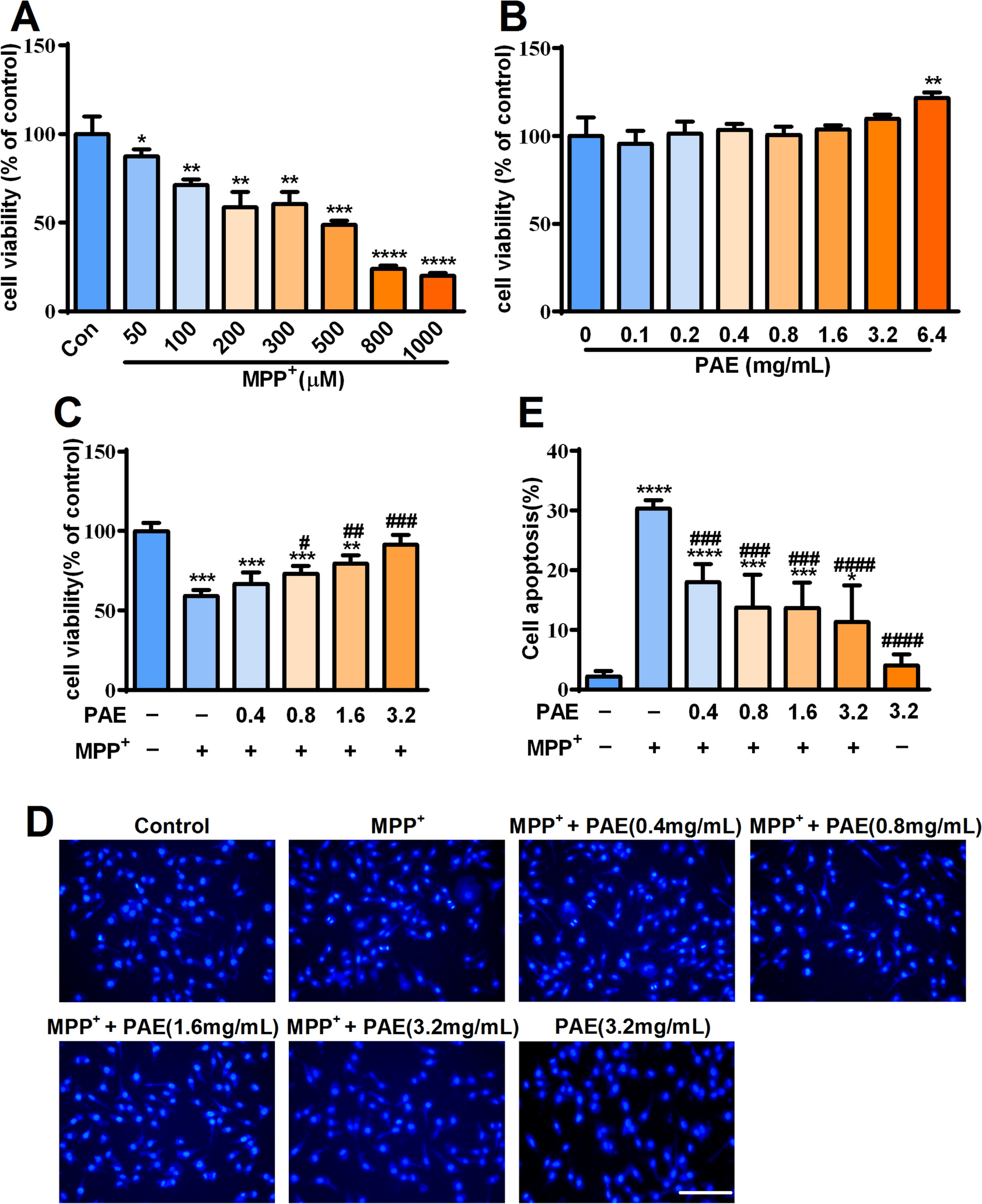
Efficacy verification of CKXR-related components in LPS-induced cell inflammation modelAccording to our study, prunasin (H97) has an extremely high exposure in vivo. Furthermore, it has been shown that part of prunasin is metabolized from amygdalin in vivo and that amygdalin (H88) also has a relatively high exposure, which was also verified in our laboratory. Amygdalin and prunasin are the same cyanogenic glycoside analogs, and in the foregoing, we have successfully constructed a spectrum of cyanogenic glycoside-exposed components from CKXR in mice. We used AUC pooled method to analyze the related components in plasma [27, 28]. With this method, the peak area can represent in vivo exposure of the components to some extent. We added all related peak areas of cyanogenic glycosides in CKXR under positive ion mode, and found that they accounted for 43.16% of the whole peak areas. Similarly, we added all related peak areas of cyanogenic glycosides in CKXR under negative ion mode, and found that they accounted for 53.57% of the whole peak areas. Relevant studies have reported that amygdalin has definite anti-inflammatory effects [25]. Prunasin has a higher exposure and it was interesting to see whether it possessed an anti-inflammatory effect comparable to amygdalin. We selected two cyanogenic glycosides, amygdalin and prunasin, which have been successfully structurally identified, and also set up the group of CKXR as a whole to initially investigate their anti-inflammatory activities. Based on the concentration of amygdalin in CKXR, we chose the concentration of 10 mg/mL of CKXR medicinal solution and 6.25 μmol/L of amygdalin, in which the concentration of amygdalin was comparable. The concentration setting of prunasin was consistent with that of amygdalin.
qRT-PCR. Figure 5A showed that the model group could induce a significant increase in the mRNA expression of pro-inflammatory cytokines IL-1β, IL-6 and TNF-α in RAW264.7 cells after 24 h of LPS stimulation compared with the normal control group, and the differences were all statistically significant (p < 0.001, p < 0.0001, p < 0.01). The mRNA expression of IL-1β, IL-6 and TNF-α was significantly down-regulated in all groups after the intervention with amygdalin, prunasin and CKXR compared to the model group. The differences in IL-1β and IL-6 were statistically significant in the CKXR, amygdalin and prunasin groups compared to the model group (p < 0.001, p < 0.0001). However, down-regulation of TNF-α by CKXR (p < 0.01) was more significant than that of amygdalin and prunasin groups (p < 0.05).
Fig. 5
Effects of amygdalin, prunasin and CKXR on the expression of IL-1β, IL-6 and TNF-α in LPS-induced RAW264.7 cell inflammation model. A Effect of mRNA expression level; B Effect of release level. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
ELISA. Figure 5B shows that the model group induced a significant increase in the expression of pro-inflammatory cytokines IL-1β, IL-6 and TNF-α in RAW264.7 cells after 24 h of LPS stimulation compared with the normal control group, and the differences were all statistically significant (p < 0.05, p < 0.0001, p < 0.0001). Compared with the model group, the expressions of IL-1β, IL-6 and TNF-α were all down-regulated after the administration of the intervention, although their down-regulation of IL-1β was not significant. However, the down-regulation of both IL-6 and TNF-α by CKXR was statistically significant (p < 0.0001, p < 0.0001).
Combining the above qRT-PCR and ELISA results, we found that both of amygdalin and prunasin have anti-inflammatory effect, in which the anti-inflammatory effect of prunasin is superior to that of amygdalin at the same concentration. This suggests that amygdalin and prunasin are most likely dependent on the consistent parent structure of both in exerting their anti-inflammatory effects. For prunasin, only one glycoside is linked to the parent structure, and its affinity for potential targets may be stronger than that of amygdalin. In addition, we found that when the CKXR administration solution was compared with the amygdalin group at comparable concentrations, the anti-inflammatory activity of the monomer was not superior to that of the overall. This suggests that the anti-inflammatory effect of CKXR is not solely exerted by amygdalin, but other cyanogenic glycosides (including prunasin) also play an important role and deserve further attention. The specific mechanism of superimposed or synergistic effects of different cyanogenic glycosides can be further verified in the future.
留言 (0)