
記住我
Plant productivity on a global scale faces a significant threat from heat stress, leading to notable reductions in crop yields. The adverse impacts of heat stress on the growth and development of plants are varied and can manifest at different stages of their life cycle (Zhao et al., 2020). In particular, legumes seem sensitive to heat stress during the blooming stage. Exposure to elevated temperatures ranging from 30°C to 35°C for a few days can severely impact key reproductive processes, including microsporogenesis, megasporogenesis, fertilization, and early embryogenesis. This can result in damaged pods and significant losses in yield (Mohapatra et al., 2020; Liu et al., 2019). Furthermore, heat stress negatively impacts various physiological aspects, including photosynthesis, respiration, membrane stability, the production of reactive oxygen species (ROS), and the accumulation of antioxidants (Djanaguiraman et al., 2020; Hasanuzzaman et al., 2013). It is important to note that heat stress is often compounded by other abiotic stresses like salt or drought, but understanding the independent mechanisms by which heat stress affects plant growth and metabolism is crucial for effectively mitigating the combined effects of these stresses.
Plants have developed a range of mechanisms to confront heat stress, allowing them not only to survive but also thrive in high-temperature surroundings. These mechanisms can be broadly classified into avoidance and tolerance strategies (Bita and Gerats, 2013; Cao et al., 2023). The specific coping mechanisms of a plant depend on its species and the prevailing environmental conditions. In response to heat stress, plants exhibit distinctive cellular and molecular reactions involving alterations in cell structure organization and membrane functions aimed at enduring challenging conditions. This period triggers an up-regulation of genes responsible for molecular chaperones and signaling molecules that orchestrate events in response to heat stress (Boopathy et al., 2022; Ul Haq et al., 2019).
One particular set of co-chaperones involved in these responses are BAG (B-cell lymphoma-2 associated athanogene) proteins, which regulate processes like protein folding, cellular signaling, translocation, and degradation during heat stress (Arif et al., 2021; Nawkar et al., 2017). BAG proteins are evolutionarily conserved, showcasing multifunctional properties that contribute to cell survival in scenarios such as apoptosis and stress responses (Kabbage and Dickman, 2008; Wang et al., 2020; Huang et al., 2022). A distinctive feature of BAG proteins is the conserved BAG domain (BD) located at their C-terminus, which interacts with the ATPase domain of heat shock protein 70 (HSP70). HSP70 is a molecular chaperone critical for folding and unfolding proteins, playing a pivotal role in shielding cells from damage (Doukhanina et al., 2006).
Within the plant kingdom, the BAG protein family can be categorized into two groups based on their structural characteristics, determined through genome-wide alignment and conservative domain identification (Jiang et al., 2023; Kabbage and Dickman, 2008). The first group possesses an N-terminal ubiquitin-like (UBL) domain, while the second group features a plant-specific CaM-binding IQ motif near the BAG domain (Doukhanina et al., 2006; Dash and Ghag, 2022). Despite the known roles of BAG proteins in responding to abiotic and biotic stresses in plants, limited research has explored their specific involvement in the response to heat stress.
The AtBAG4 gene in Arabidopsis exhibited increased expression in response to heat stress, and it subsequently interacted with HSP70 to prevent cell death induced by the stress. A heat-shock element within the AtBAG5 promoter was also identified to respond to heat induction (Nawkar et al., 2017). In maize, heat stress led to the upregulating of 13 genes belonging to the BAG family (Li-Zong et al., 2013). The potential BAG gene, HSG1, in grapes, demonstrated heat shock-inducible properties. The overexpression of HSG1 in Arabidopsis resulted in enhanced heat tolerance, even at extremely high temperatures (Kobayashi et al., 2012). Additionally, TaBAG2, identified as a stress-inducible gene in wheat, conferred thermotolerance when overexpressed in Arabidopsis (Ge et al., 2016). On the contrary, the BAG gene from tomato, specifically the SlBAG9 gene, which is a homolog of AtBAG5, exhibited negative regulation concerning heat tolerance. Overexpressing SlBAG9 led to the development of tomato plants that are highly sensitive and susceptible to heat stress (Ding et al., 2022).
Pigeonpea (Cajanus cajan) is a leguminous crop known for its high protein content. It is a significant provider of essential nutrients such as proteins, vitamins, and minerals in human diets and cattle feed (Kuraz Abebe, 2022). However, the productivity of this crop has stagnated in recent decades due to the detrimental impact of abiotic stresses in the evolving climate. The cumulative consequences of heat stress on pigeonpea result in diminished yield and altered crop quality. The degree of yield reduction varies depending on factors such as the growth stage, intensity, and duration of heat stress. Although the involvement of BAG genes in responding to heat stress has been documented in various plants like Arabidopsis (Doukhanina et al., 2006), tomato (Ding et al., 2022), wheat (Ge et al., 2016), and maize (Li-Zong et al., 2013), there is currently a lack of comprehensive genome-wide phylogenetic and functional studies on BAG genes specifically for pigeonpea. However, the recent availability of high-quality genomic data and the increasing research focus on climate change, particularly heat stress, have highlighted significant environmental challenges that impact pigeonpea growth and production. These developments have prompted us to prioritize functional genomic studies, including identifying gene families such as BAG, which are known to be involved in heat stress responses in Arabidopsis.
To gain a deeper insight into the evolutionary dynamics of BAG genes in pigeonpea and uncover their potential contributions to heat stress response, we identified nine CcBAG genes in pigeonpea. Through in silico analyses, we documented various aspects, including their chromosomal locations, motif compositions, cis-acting elements, evolutionary relationships, genomic collinearity, and selection pressure. Additionally, we conducted an expression analysis of these BAG genes under heat stress conditions. These findings offer valuable insights into the functions of BAG family members and present opportunities for utilizing them in the development of heat-tolerant pigeonpea varieties.
Materials and methodsIdentification and sequence analysis of the pigeonpea BAG gene familyTo identify BAG genes in pigeonpea, an exhaustive search was conducted using the coding sequences of seven BAG genes from Arabidopsis thaliana, sourced from The Arabidopsis Information Resource (TAIR) at (https://www.arabidopsis.org/). These genes served as blast queries applied to the pigeonpea genomic sequence data available on the Legume Information System (LIS) database (https://www.legumeinfo.org/). Using the physical location information retrieved from the LIS database, all identified pigeonpea BAG genes were subsequently mapped to chromosomes with the assistance of the MapGene2Chromosome (MG2C) tool (Version 2.1, accessible at (http://mg2c.iask.in/mg2c_v2.1/). The molecular weight, theoretical isoelectric point (pI), aliphatic index and grand average of hydropathicity (GRAVY) values of the BAG genes were extracted using the Expasy protparam tool (https://web.expasy.org/protparam/) (Gasteiger et al., 2005). The in silico analysis workflow for the pigeonpea BAG gene family is summarised in Supplementary Figure S1.
Phylogenetic analysis and sub-cellular localizationA total of 49 amino acid sequences representing BAG family proteins from C. cajan, Glycine max, Medicago truncatula, Arachis hypogea, and Cicer arietinum were extracted from the LIS database using the coding sequences of the BAG genes from A. thaliana as a template. The amino acid sequences of Arabidopsis were retrieved from the TAIR database. These amino acid sequences underwent alignment through the MUSCLE algorithm, and a phylogenetic tree was constructed using MEGA 11.0, employing the neighbour-joining method and a bootstrap test involving 1,000 replications (Tamura et al., 2021). Additionally, the subcellular localization of pigeonpea BAG family proteins was predicted in silico using the BUSCA tool, accessible at (https://busca.biocomp.unibo.it/) (Savojardo et al., 2018).
Domain analysis, gene structure analysisBAG-conserved domains were generated using the HMMER web server (Bio sequence analysis using profile hidden Markov Models) (Potter et al., 2018) (https://www.ebi.ac.uk/Tools/hmmer/). The presence of these domains in the BAG proteins was further validated using the Interpro scan tool (https://www.ebi.ac.uk/interpro/search/sequence/). To visualize the exon-intron structures of the BAG genes, coding and genome sequences were employed in conjunction with the Gene Structure Display Server software (http://gsds.gao-lab.org/) (Hu et al., 2015).
Collinearity and evolutionary analysis of identified BAG genesMedicago truncatula, recognized as a model legume plant, displays a substantial abundance of widely conserved regions with pigeonpea, surpassing other legumes. Due to these distinctive characteristics, M. truncatula was selected as the model for conducting genome collinearity and evolutionary analyses of BAG genes in pigeonpea. The analysis was carried out using the Multiple Collinearity Scan toolkit (MCScanX) program integrated into TBtools, with specific parameters set as follows: CPU for BLASTp (2), Number of BLAST hits (5), and E-value (1e−10). The multiple synteny plotter tool within TBtools was employed to identify orthologous genes in the organisms, visually connecting orthologous BAG genes of pigeonpea with M. truncatula using curved arches. To determine the non-synonymous substitution rates and synonymous substitution rates of the identified orthologous gene pairs, the Ka/Ks calculator tool in TBtools version 2.0 was utilized (Chen et al., 2023).
Cis-regulatory element analysis of the identified BAG genesTo identify the cis-regulatory elements within the promoter region of the identified BAG genes, a 2,000-bp upstream region from the transcriptional start site for each gene was acquired from Cajanus Mine (https://mines.legumeinfo.org/cajanusmine/begin.do). The identification of cis-regulatory elements involved, submitting the obtained upstream sequences to the PlantCARE online tool (Lescot et al., 2002) (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/). The visualization of the results from the cis-regulatory element analysis was conducted using TBtools.
Protein-protein interaction analysis and 3D structure prediction of CcBAG proteinTo predict the potential protein-protein interaction (PPI) network, the identified pigeonpea BAG proteins were submitted to the STRING online tool (Szklarczyk et al., 2015) (http://string-db.org). By utilizing BLAST, the orthologs of these proteins were recognized in tomato, and the ortholog with the most substantial bit score was subsequently examined in greater detail. Any predicted proteins that did not exhibit interactions were excluded from further consideration. Understanding the 3D structure of proteins helps us to gain insights into their functions, mechanisms, and interactions. Hence, the 3D structure of CcBAGs was predicted by the PHYER2 server (Lovell et al., 2002) (www.phyre2/html). Moreover, the I-TASSER server (Zhang, 2008) was used to verify the models. These structures were further visualized through the UCSF Chimera visualization tool (Pettersen et al., 2004).
GO and miRNA prediction of CcBAG genesTo explore the potential functions of candidate target genes, BLAST2GO (https://www.blast2go.com/) analysis was performed. The GO annotations were categorized into three distinct groups: cellular component, molecular function, and biological process. Also, the miRNA sequences for the BAG genes were obtained from the Plant microRNA Encyclopedia (PmiREN) database (https://pmiren.com/database). To identify the target sites of the retrieved miRNA sequences in these genes, psRNATarget (Dai et al., 2018) (https://www.zhaolab.org/psRNATarget/) was utilized. The visualization of the predicted target sites was then done using the Geneious Prime software package v2024.0.
Expression analysis of BAG genes during terminal heat stressPlant materialThe selfed progenies of two medium-duration pigeonpea (ICPL 87119 and ICPL 85063 - used as heat susceptible genotypes) and one mid-early genotype (TS3R-used as heat tolerant genotype) were used in this study. Pigeonpea, being inherently sensitive to both photoperiod and temperature, served as the subject of the study. To assess sensitivity to photoperiod and thermotolerance, two sets of progenies were utilized. One set consisted of flowering progenies from each genotype, while the other set comprised non-flowering progenies from the same genotypes during the summer of 2023. Notably, ICPL87119 and ICPL85063 are recognized for their heightened sensitivity to both photoperiod and temperature. In contrast, TS3R demonstrates a high level of tolerance to heat stress, particularly during the summer season.
The pigeonpea genotypes were cultivated in the deep black soils at the International Crops Research Institute for the Semi-Arid Tropics (ICRISAT) in Patancheru, Hyderabad, India (Latitude: 17.51°N, Longitude: 78.27°E, Altitude: 545 m). Planting commenced on 27 January 2023. Due to their photosensitivity, some progenies of ICPL 87119, ICPL 85063, and TS3R did not flower. On 21 May 2023, at 2 p.m., we collected flowers and leaves from the flowering plants, as well as leaf samples from the non-flowering plants. These samples were collected under a maximum recorded temperature of 40°C (Supplementary Table S1), with the plants having been exposed to temperatures ranging from 35°C to 40°C for 40–45 days. These samples were then used for expression analysis.
Gene expression analysisFor the quantitative reverse-transcription polymerase chain reaction (qRT-PCR), we isolated total RNA from flower and leaf samples of divergent pigeonpea genotypes exposed to heat stress using the RNeasy Plant Mini kit (Qiagen, Tokyo, Japan) with three replications. A 2.0 μg portion of purified RNA underwent cDNA synthesis following the recommended protocol (Thermoscript RT-PCR system, Invitrogen, Carlsbad, CA, United States). The quantitative PCR was performed using gene-specific primers (Supplementary Table S2) on a CFX96TM Real-Time System (Bio-Rad, India). To normalize cycle threshold (Ct) values, the pigeonpea GAPDH (XM_020358085) gene served as the housekeeping gene (Sinha et al., 2015). The qRT-PCR reaction conditions were set in triplicates as follows: 2 min at 95°C, 40 cycles of 10 s at 95°C, and 20 s at 60°C. Additionally, a fluorescence measurement at each 0.5°C variation from 60°C to 95°C over 20 min was included to obtain the melting curve. The relative fold expression was calculated using the 2−ΔΔCt method (Livak and Schmittgen, 2001).
ResultsIdentification and sequence analysis of the pigeonpea BAG gene familyThe Arabidopsis BAG genes were used as query sequences to search for their orthologs against the pigeonpea in the LIS database (Nawkar et al., 2017). A total of 12 genes were initially retrieved, and subsequent analysis using InterProScan and manual inspection led to the removal of redundant or false-positive genes. Ultimately, nine non-redundant CcBAG genes were identified in pigeonpea, comprising 2 BAG1 (Cc_18023 and Cc_26867), 1 BAG2 (Cc_19692), 4 BAG4 (Cc_15012, Cc_11905, Cc_01922, and Cc_16501), 1 BAG5 (Cc_19448), and 1 BAG6 (Cc_02358).
These nine CcBAG proteins exhibited molecular weights ranging from 19.41 to 129.75 kDa, and their corresponding amino acid lengths varied between 172 and 1,156 (Supplementary Table S3). The predicted theoretical isoelectric points (pI) for Cc_15012, Cc_01922, Cc_16501, Cc_19448, and Cc_02358 proteins fell within the acidic range, while the pI for the remaining proteins (Cc_18023, Cc_26867, Cc_19692, and Cc_11905) were in the basic range. The aliphatic index ranged from 55.47 to 101.56 indicating the thermostability of these CcBAG proteins. It was 101.56 for the gene Cc_19448 predicting it to be the most thermostable, while Cc_02358 identified to be the least with a value of 55.47. Further, the GRAVY values predicted for these BAG genes were all negative ranging from −0.062 (Cc_19448) to −1.092 (Cc_02358), indicating those to be hydrophilic. Additional important properties, including the length of the coding region (CDS), the number of exons/introns, chromosomal location, and cellular localization of the identified genes, were investigated to enhance our understanding of BAG genes in pigeonpea (Supplementary Table S3).
The CcBAG genes identified were spread across five chromosomes in pigeonpea: Chr01, Chr05, Chr07, Chr08, and Chr11. Notably, Chr08 contained three distinct CcBAG genes (Cc_18023, Cc_19448, Cc_19692). Chr01 included the genes Cc_01922 and Cc_02358, Chr07 had Cc_15012 and Cc_16501, and Chr11 and Chr05 housed the genes Cc_26867 and Cc_11905, respectively (Figure 1).
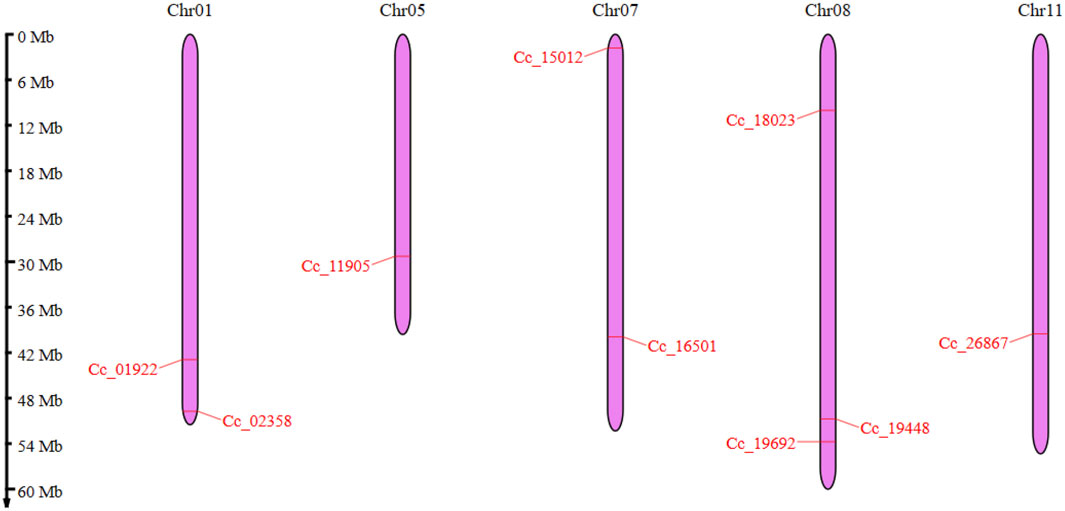
Figure 1. Chromosomal locations of BAG genes in pigeonpea. The chromosomes are represented in purple color and the genes are shown in red colored lines. The Scale on the left side represents the length of the chromosome in Mb.
Phylogenetic analysis and sub-cellular localizationTo identify the phylogenetic relationships among pigeonpea BAG proteins implicated in the regulation of heat stress, a neighbour-joining phylogenetic tree was constructed. This analysis encompassed BAG proteins from C. cajan (9 genes), A. thaliana (7 genes), G. max (12 genes), M. truncatula (5 genes), A. hypogea (10 genes), and C. arietinum (6 genes) (Figure 2). In total, 49 genes were organized into five distinct clades labelled as I to V. Among these clades, clade II exhibited the highest membership with 18 members, followed by clade I with 10 members, and clades III, IV, and V, each comprising 7 members.
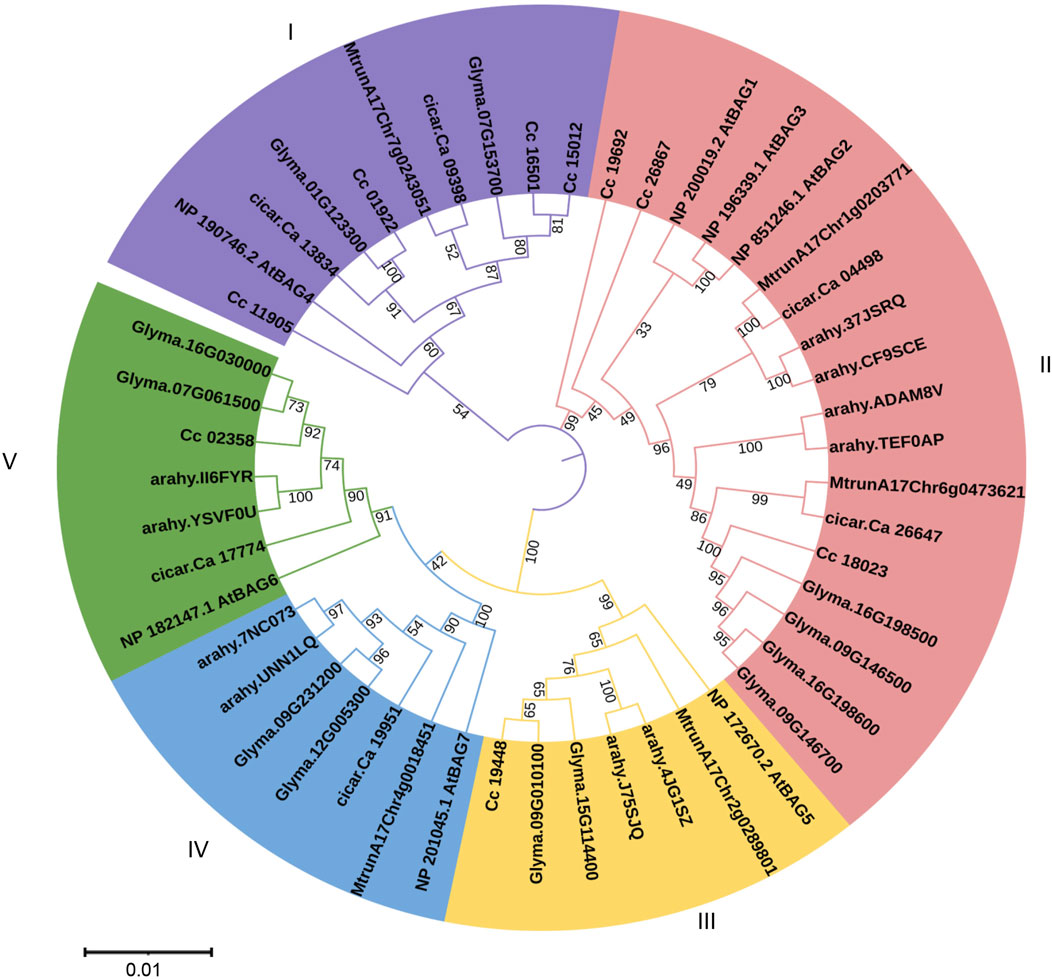
Figure 2. Phylogenetic analysis of BAG genes of Arabidopsis, pigeonpea, chickpea, soybean, medicago, and groundnut. Five clades are represented in five colors. Trees are constructed using the Neighbor-Joining method with 1,000 bootstrap replications in MEGA X software.
The CcBAG proteins were examined for their anticipated subcellular localization using the BUSCA (Bologna Unified Subcellular Component Annotator) tool. The outcomes revealed that out of the nine proteins, seven CcBAGs were predicted to be localized in the nucleus, with the exception of Cc_26867 (BAG1) and Cc_19448 (BAG5), which were predicted to be situated in the chloroplast (Supplementary Table S3).
Collinearity and evolutionary analysis of identified BAG genesTo better understand the phylogenetic connections among pigeonpea BAG genes and other members of the Fabaceae family, we conducted a collinearity analysis involving M. truncatula (Figure 3). Among the 9 CcBAG genes identified in pigeonpea, only 7 exhibited collinear genes in M. truncatula, with Cc_26867 and Cc_11905 lacking collinear counterparts in the considered species.
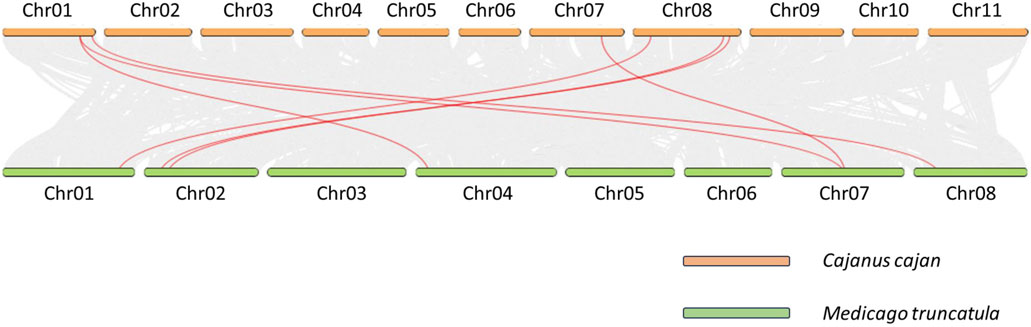
Figure 3. Collinearity analysis of BAG genes. Orthologous collinear BAG genes of Cajanus cajan and Medicago truncatula are represented by red curved lines on a gray background.
The identification of the synonymous to non-synonymous mutation ratio (Ka/Ks) aids in understanding the selection pressure acting on identified gene pairs in pigeonpea (Supplementary Table S4). Among the 9 genes, only 6 (Cc_18023, Cc_01922, Cc_15012, Cc_16501, Cc_19448, and Cc_02358) displayed significant collinearity with Medicago and were subsequently subjected to Ka/Ks analysis (Wang et al., 2013; Hyun et al., 2014). The calculated Ka/Ks values ranged from 0.07 to 0.45, all of which are less than 1. This suggests that these six CcBAG genes underwent purifying or stabilizing selection pressure throughout their evolutionary history.
Domain analysis and gene structure analysisThe BAG domain (BD) located in the C-terminus of the genes was identified using the HMMER web server. Additionally, the genes Cc_18023 (BAG1), Cc_19692 (BAG2) in group II, and Cc_11905 (BAG4) in group I were found to contain UBL domains in addition to the BAG domain. Conversely, Cc_02358 (BAG6) in group V featured an IQ calmodulin-binding motif situated in the N-terminus of the gene (Figure 4). In the case of CcBAGs, the number of exons ranged from 2 to 4. All genes in Clades I and II possessed four exons, while those in Clades III and V had only two exons (Figure 5). These findings suggest a high level of evolutionary conservation among members of the pigeonpea BAG gene family.
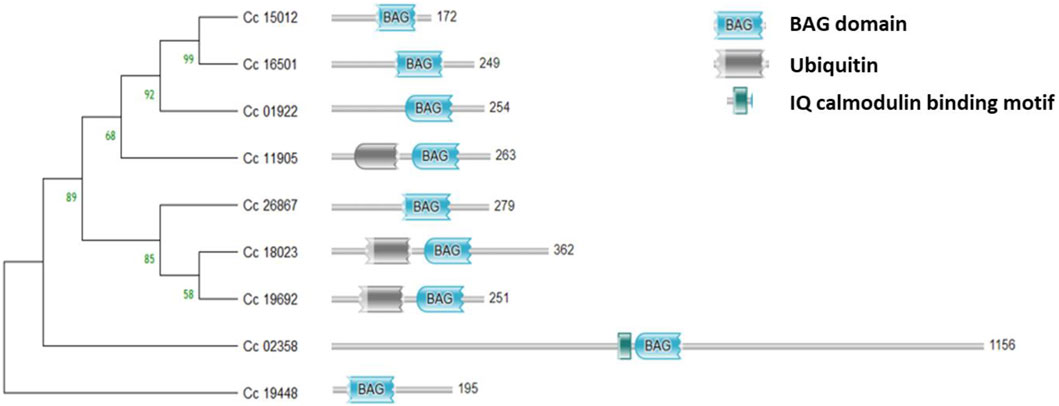
Figure 4. Domain analysis of the identified BAG genes showing the conserved BAG domain in the C-terminus, Ubiquitin domain in the N-terminus, and IQ calmodulin-binding domain.
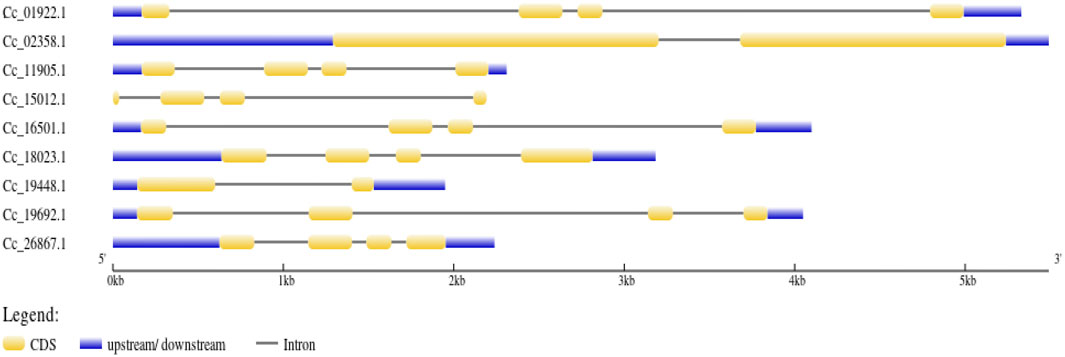
Figure 5. Gene structure representation of BAG genes. Exons are represented as yellow boxes; introns are represented as black lines while untranslated regions (UTRs) are represented as blue boxes.
Cis-regulatory element analysis of the identified BAG genesTo further analyze the potential regulatory mechanisms of CcBAG genes, cis-elements associated with the putative promoter regions of these genes were identified by extracting the 2,000 bp upstream sequence from the transcription start site and analysed for cis-regulatory elements (Figure 6).
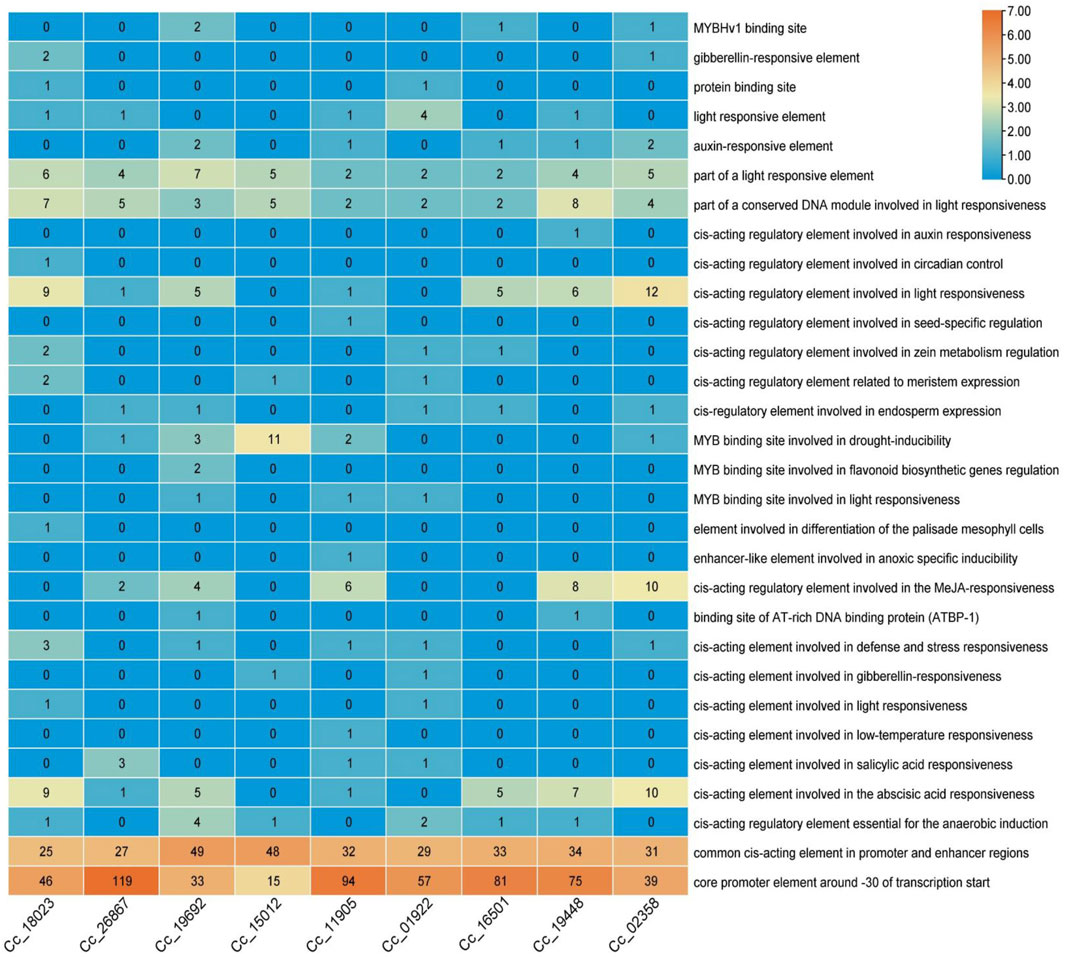
Figure 6. Cis-regulatory elements analysis of BAG genes. The scale on the right represents the color scale based on the number of cis-regulatory elements.
The findings revealed the presence of shared upstream elements responsive to various environmental stimuli, such as light, defense, stress, low-temperature, and drought. Specifically, cis-acting elements involved in defense and stress responsiveness, cis-acting elements involved in low temperature responsiveness are observed, with elements for light responsiveness noted high in BAG5 (Cc_19448), and 1 BAG6 (Cc_02358). Additionally, elements associated with anaerobic and anoxic induction, along with different binding sites for AT-rich DNA proteins and MYB (myeloblastosis-related proteins) involved in drought inducibility caused by heat stress that are specifically high in BAG4 (Cc_15012), were identified. Furthermore, several elements responsive to hormones like gibberellin, auxin, methyl jasmonic acid (MeJA), abscisic acid, and salicylic acid, as well as growth-related elements like circadian control, endosperm expression, and elements involved in the differentiation of palisade mesophyll cells were observed. In particular, elements involved in MeJA responsiveness and abscisic acid responsiveness, which are possible hormone-regulated pathways in heat stress mechanism, are seen exceptionally in BAG5 (Cc_19448) and 1 BAG6 (Cc_02358). The occurrence and quantity of these elements in the upstream regions of the identified genes varied among different genes. Notably, among all the elements present in the upstream regions of nearly all CcBAG genes, light-responsive elements constituted more than half of the identified elements.
Protein-protein interaction analysis and 3D structure prediction of CcBAG proteinsThe study of protein–protein interactions is crucial as it provides insights into the molecular regulatory network. In this particular investigation, the interaction network of CcBAG proteins revealed their potential interactions with multiple proteins. Notably, proteins of Cc_18023, Cc_15012, Cc_11905, and Cc_01922 did not exhibit interactions with other proteins. On the other hand, proteins of Cc_19448, Cc_26867, Cc_19692, Cc_02358, and Cc_16501 displayed interactions both among themselves and with additional proteins (Figure 7). This interaction network highlighted A0A3Q7I6P6 (HSP70) as the primary functional node with multiple protein interactions. This suggests that the HSP70 protein interacts with the majority of CcBAG proteins and is likely activated in response to defense and stress conditions (Supplementary Table S5). The knowledge of protein 3D structure acts as a pre-requisite to understanding the function of a protein, as a small variation in the sequence of protein could lead to greater structural variation than the native protein. The 3D protein structures of the CcBAG genes revealed that the BAG1 (Cc_18023 and Cc_26867), BAG2 (Cc_19692) and BAG4 (Cc_15012, Cc_11905, Cc_01922, and Cc_16501), displayed almost identical structures. The helices, turns/Beta strands and loops were seen very similar in all their structures. The predicted similar structures indicate that these BAG proteins may have potentially similar functions. In contrast, the genes BAG5 (Cc_19448) and BAG6 (Cc_02358) each exhibited significantly different structures with long helices and almost no turns/Beta strands (Supplementary Figure S2).
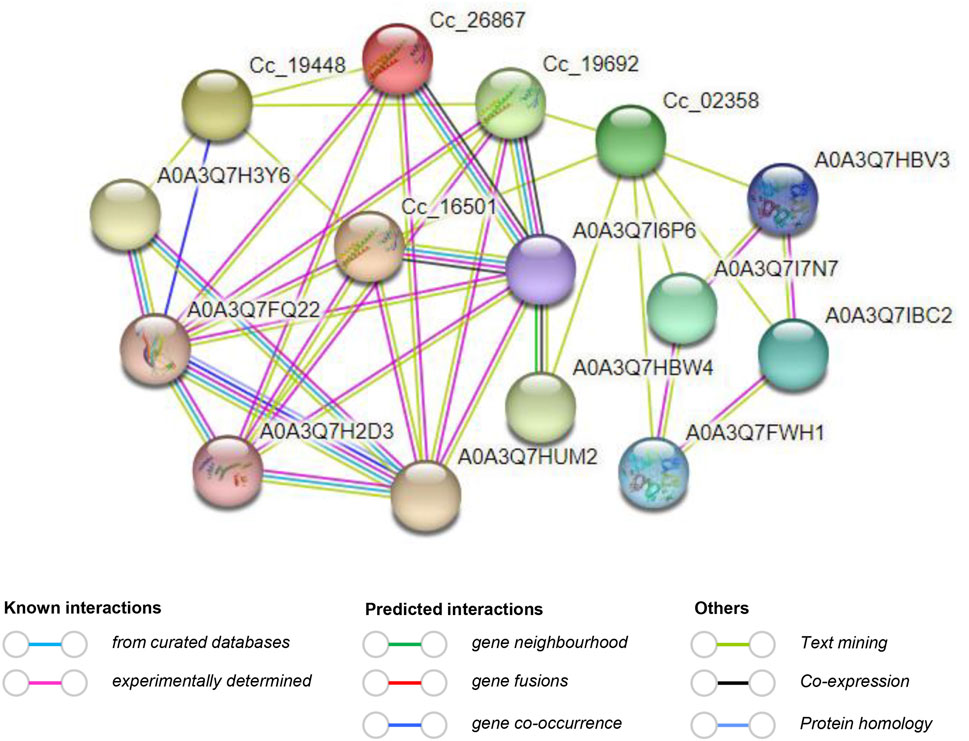
Figure 7. Predicted protein-protein interaction network of CcBAG proteins obtained from STRING (https://string-db.org/) database. The red line indicates the presence of fusion evidence; The green line is neighborhood evidence; The blue line is co-occurrence evidence; The purple line is experimental evidence.
GO and miRNA prediction of CcBAG genesThe GO enrichment analysis facilitates a better understanding of different functional processes involving CcBAG gene family members. It was revealed that all nine BAG genes were involved in the cellular anatomical entity (GO: 0005575) of the cellular component category, and binding (GO: 0003674) of the molecular function category (Supplementary Figure S3). We got 7 of the BAG gene sequences that are involved in biological regulation (GO:0008150); 3 sequences were identified for cellular process (GO: 0008150) and metabolic process (GO: 0008150); 1 sequence for response to stimulus (GO: 0008150) in biological process category. Similarly, for the molecular function category, 7 of the CcBAG sequences are involved in molecular function regulatory activity (GO: 0003674), and 1 of the sequence for catalytic activity (GO:0003674) were observed. In cellular component category, the intracellular anatomical structure (GO: 0110165) and cytoplasm (GO: 0110165, GO:0005622) were controlled by 7 of the CcBAG sequences and 2 of the sequences were shown for the remaining cellular components. The GO enrichment analysis reveals that, out of the 9 CcBAG genes, 7 exhibit similar biological, cellular, and molecular functions, indicating their conserved nature compared to Arabidopsis throughout evolution. In contrast, two BAG genes—likely BAG5 (Cc_19448) and BAG6 (Cc_02358)—show divergence in their biological, cellular, and molecular functions. This divergence is supported by differences in their protein 3-dimensional structures, which may contribute to their altered functions.
The miRNAs control/regulate gene expression at the posttranscriptional level by base pairing with complementary mRNA sequences, leading to gene silencing or degradation of the target mRNA under several stress-related conditions. Hence, the CcBAG genes targeted by the miRNAs have been predicted. Out of the nine BAG genes, only Cc_02358 (BAG6) gene was found to be targeted by two different miRNA accessions (miRN5120 and miRN5144) with perfect complementarity (Supplementary Figure S4), leading to translational inhibition and silencing of the BAG gene irrespective of its higher expression in susceptible genotypes and different protein 3-dimensional structure that facilitates the post translational inhibition.
Differential expression of BAG genes in pigeonpea during heat stressTo elucidate the expression patterns of the CcBAG genes, flower and leaf tissues from diverse pigeonpea genotypes exposed to heat stress were chosen. Following established practices, the GAPDH gene, a potentially functional endogenous reference gene commonly utilized for normalization, was selected based on prior studies (Sinha et al., 2015). While all nine CcBAG genes were expressed in all the genotypes, it was evident that each CcBAG gene exhibited a distinct expression pattern (Figure 8; Supplementary Figure S5).
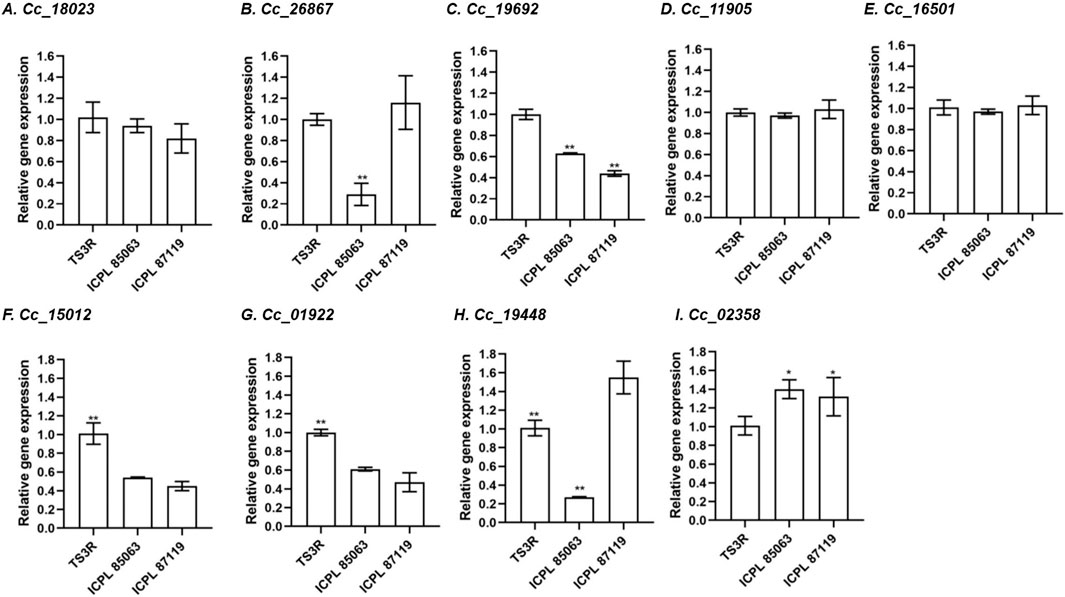
Figure 8. Relative transcript expression in flower tissues of contrasting pigeonpea genotypes during heat stress, based on qRT–PCR in comparison to reference gene GAPDH. (A). Cc_18023 (BAG1), (B). Cc_26867 (BAG1), (C). Cc_19692 (BAG2), (D). Cc_11905 (BAG4), (E). Cc_16501 (BAG4), (F). Cc_15012 (BAG4), (G). Cc_01922 (BAG4), (H). Cc_19448 (BAG5), (I). Cc_02358 (BAG6). TS3R is a heat-tolerant genotype; ICPL 85063 and ICPL 87119 are heat-susceptible genotypes. *P< 0.05, **P< 0.01, a significant difference in expression level in tolerant compared to susceptible ones.
In flower tissues, the heat-tolerant genotype TS3R exhibited upregulation of four genes, namely Cc_18023 (BAG1), Cc_19692 (BAG2), Cc_15012, and Cc_01922 (BAG4) compared to susceptible genotypes. Two genes, Cc_26867 (BAG1) and Cc_19448 (BAG5), were exclusively downregulated in the ICPL 85063 genotype. Conversely, one gene Cc_02358 (BAG6), exhibited upregulation in the susceptible genotypes (ICPL 85063 and ICPL 87119), indicating that this gene, along with Cc_18023 (BAG1), Cc_19692 (BAG2), Cc_15012 and Cc_01922 (BAG4), might play a crucial role in the regulation of heat stress.
Regarding leaf tissue in flowering plants, the gene Cc_02358 showed notable upregulation in the heat-tolerant genotype TS3R. Conversely, both Cc_02358 and Cc_11905 exhibited significant upregulation in the susceptible genotype ICPL 87119. In contrast, when it comes to leaves of non-flowering plants, all CcBAG genes displayed similar expression levels across the three genotypes, regardless of their tolerance or susceptibility to heat stress (Supplementary Figure S5).
DiscussionThe BAG gene family represents a set of highly conserved proteins found across all eukaryotes, including plants. These proteins play crucial roles in diverse cellular processes such as stress response, development, and immunity (Huang et al., 2022; Jin et al., 2015; Jiang et al., 2023; Dash and Ghag, 2022). Extensive characterization of the BAG gene family has been conducted in model plants like Arabidopsis (Doukhanina et al., 2006), rice (Rana et al., 2012), wheat (Ge et al., 2016), and soybean (Wang et al., 2020). However, the BAG genes in pigeonpea have not been characterized to date. In this study, we identified the BAG genes in pigeonpea, explored their evolutionary relationships, compared their sequence features, and scrutinized their expression patterns in flower and leaf tissues under terminal heat stress.
Identification, evolution, and characteristics of the BAG genes in pigeonpeaThrough sequence analysis in the CajanusMine database, we identified nine BAG genes in pigeonpea, and these genes are distributed across five chromosomes. Pigeonpea exhibits a smaller number of BAG genes compared to other legume plants like soybean (12) and groundnut (10), but a higher count than Medicago (5) and chickpea (6). This observation suggests that in dicots, the BAG protein family may be primarily associated with lineage-specific Whole Genome Duplication (WGD) events (Young et al., 2011; Schmutz et al., 2010). The genes of BAG1, BAG2 and BAG4 featured UBL domain, and BAG6 showed the presence of calmodulin-binding IQ motif that binds to calmodulin (CaM), as reported previously by Doukhanina et al. (2006); Nawkar et al. (2017); Kabbage and Dickman (2008).
A phylogenetic analysis of the BAG protein family, categorizing members into five clades, aligns with similar patterns observed in tomato (Irfan et al., 2021) and soybean (Wang et al., 2020). In contrast, groundnut and M. truncatula have reported four clades. These findings suggest that the preservation of specific clades may vary among species due to their distinct evolutionary histories. Moreover, events involving the gain or loss of exons/introns contribute to gene structure divergence and functional differentiation (Xu et al., 2012). Understanding the exon-intron structure is crucial for unraveling the relationships between evolutionary processes and functional divergence (Cao et al., 2016; Moore and Purugganan, 2003; Hurley et al., 2005).
In the pigeonpea BAG gene family, the number and length of exons/introns exhibit non-conservation, a trend shared with other species such as Arabidopsis (Doukhanina et al., 2006), banana (Dash and Ghag, 2022), rice (Rana et al., 2012), wheat (Ge et al., 2016), and soybean (Wang et al., 2020). These differences in exon/intron characteristics may arise from chromosome rearrangements and fusions, potentially leading to distinct biological functions for these genes.
To systematically explore the overall organization of members and establish connections between proteins, biological functions, and subcellular localization, it is crucial to investigate how proteins are distributed within a cell. Subcellular localization aims to assign proteins, responsible for specific biological functions, to specific cell compartments. For example, all CcBAGs predominantly localize in the nucleus, except for Cc_26867 (BAG1) and Cc_19448 (BAG5), which are predicted to be located in the chloroplast. This differs from the reported localization of Arabidopsis BAG proteins, where AtBAGs 1-3 were found in the cytoplasm and AtBAG4 in both the cytoplasm and nucleus (Nawkar et al., 2017). AtBAG5 and AtBAG6 were reported to localize in the mitochondria and vacuole (Doukhanina et al., 2006). This disparity could be attributed to potential nuclear localization signals that are not deposited in the databases, indicating the existence of such signals in BAG proteins (Irfan et al., 2021).
Regulation of BAG genes in pigeonpeaPromoter regions of the CcBAG genes were found to contain putative cis-acting elements associated with light, stress, hormones, and development, suggesting potential regulation by various abiotic stress factors and hormones. The identification of conserved stress-responsive cis-elements such as Long Terminal Repeats (LTR), MYB binding sequences (MBS), MYB-binding sites, and TC-rich repeats implies that CcBAG genes may be subject to stress regulation. This observation aligns with the enrichment of stress-responsive elements like MBS and TC-rich repeats in Arabidopsis BAG genes (Doukhanina et al., 2006; Nawkar et al., 2017). The presence of stress-related elements in the upstream regions of BAG genes in pigeonpea mirrors the pattern observed in BAG genes from other crops like Arabidopsis, rice, soybean, wheat, and tomato. Moreover, successful applications of such information for enhancing stress tolerance in rice and Arabidopsis have been reported (Arif et al., 2021; Ge et al., 2016; Irfan et al., 2021; Wang et al., 2020).
Regulatory relationship between BAGs and HSPs in pigeonpeaThe protein-protein interaction map revealed a network comprising 15 nodes and 37 edges, with an average node degree of 4.93. The genes of BAG1, BAG2, and BAG4 interacted with HSP70 protein. A similar result of A. thaliana AtBAG4, known to bind with HSP70, has been reported to suppress abiotic-induced cell death (Doukhanina et al., 2006). The PPI network further indicated that the BAG proteins which were not directly interacting with HSP70, formed complexes with the CcBAGs that did interact with HSP70. This observation suggests the formation of complexes involving molecular chaperones and signaling molecules. The results imply that BAG domains, in regulating stress responses, enable BAG-family proteins to function as adapter molecules by recruiting HSP70/HSC70 to modulate the activity of target proteins. Similar findings regarding the interaction of BAG proteins with HSPs were reported in tomato (Irfan et al., 2021).
Expression analysis of BAG genes in pigeonpea during heat stressPlants have evolved mechanisms to cope with challenging environmental conditions and withstand various stresses (Song et al., 2019). Previous research has demonstrated the involvement of BAG genes in responding to multiple stresses (Jiang et al., 2023). In our investigation of the expression of nine CcBAG genes in genotypes with varying responses to heat stress, we utilized qRT-PCR to analyze their expression in flower and leaf tissues. Significant differences were observed in the expression levels of these genes in both flowering and non-flowering leaves. Considering the severe impact of heat stress on the reproductive stage of plants, our focus was primarily on flower tissues. In flower tissues, the heat-tolerant genotype TS3R exhibited upregulation of BAG4 genes (Cc_15012 and Cc_01922). This finding aligns with a previous study (Doukhanina et al., 2006) where AtBAG4 was reported to interact with HSP70 and suppress cell death induced by abiotic stress. A significant upregulation in the expression of Cc_19448 (BAG5) in the susceptible genotype ICPL 87119 was in parallel to an experimental result in tomato where overexpression of SlBAG9 (a homologous gene of AtBAG5) exhibited high sensitivity to heat stress (Ding et al., 2020; Ding et al., 2022). Moreover, AtBAG6 is known as a heat-inducible protein (Kang et al., 2006; Li et al., 2017; Fu et al., 2019). Previous research indicated that the AtBAG6 gene responds to heat stress, and its mutation can impair plant thermotolerance (Nawkar et al., 2017). In our study, the CcBAG6 gene, Cc_02358, displayed significant upregulation in the susceptible genotypes. This observation is consistent with a study suggesting that a mutation in the AtBAG6 gene enhances basic thermotolerance in plants (Echevarria-Zomeno et al., 2016). However, a single BAG6 mutation did not confer thermotolerance (Fu et al., 2019). The knockout of AtBAG6 significantly improved the heat tolerance of the FES1A mutant (a Nucleotide Exchange Factor that increases molecular chaperone efficiency essential for heat tolerance in Arabidopsis) (Fu et al., 2019). It is also proposed that since AtBAG6 has a CaM-binding IQ motif, knocking down this gene may release CaM or CaM-like proteins from the nucleus, altering the dynamics of nuclear calcium signaling to enhance HSP expression in FES1A mutants and improve acquired thermotolerance.
Our miRNA analysis revealed that two of the identified miRNA sequences in pigeonpea perfectly complemented the CcBAG6 gene (Cc_02358) which imply either a decrease or complete loss of the protein encoded by this gene. This could likely account for the significant downregulation of this gene expression as observed in the tolerant genotype TS3R, supported by the expected gene silencing effect via translational inhibition, in miRNA target site prediction analysis.
ConclusionIn this study, we identified a total of nine pigeonpea BAG family genes along with their corresponding proteins. These genes were classified into five clades based on their phylogenetic relationships. The study delved into the gene structure, analysis of conserved domains, and an in-depth exploration of the promoter regions through in silico analysis. The findings underscore that these proteins interact with HSP70 proteins and predominantly localize in the nucleus and chloroplast. In these cellular compartments, they have the potential to regulate the activity of target proteins by forming complexes with HSP70 during plant stress responses and development. Moreover, the CcBAG genes demonstrated responsiveness to stress, with their expression patterns being modulated under heat stress conditions. The functional characterization of these CcBAG genes holds promise for a comprehensive understanding of their interactions with signaling pathways. This knowledge can contribute to precise breeding interventions aimed at developing climate-resilient pigeonpea varieties.
Data availability statementThe original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributionsCA: Data curation, Formal Analysis, Methodology, Software, Writing–original draft. AT: Data curation, Formal Analysis, Methodology, Software, Visualization, Writing–original draft. GH: Data curation, Formal Analysis, Software, Validation, Writing–original draft. NB: Formal Analysis, Methodology, Validation, Writing–original draft. PG: Methodology, Validation, Writing–review and editing. WT: Resources, Validation, Writing–review and editing. KY: Conceptualization, Funding acquisition, Investigation, Supervision, Validation, Writing–original draft, Writing–review and editing.
FundingThe author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
AcknowledgmentsPG acknowledges the funding support from Rastriya Krushi Vikas Yojana (RKVY), Government of Odisha. National Food Security Mission (NSFM), Ministry of agriculture and farmers welfare, Government of India. All the authors acknowledge funding received from the Indian Council of Agriculture Research (ICAR).
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1418380/full#supplementary-material
ReferencesArif, M., Li, Z., Luo, Q., Li, L., Shen, Y., and Men, S. (2021). The BAG2 and BAG6 genes are involved in multiple abiotic stress tolerances in Arabidopsis thaliana. Int. J. Mol. Sci. 22, 5856. doi:10.3390/ijms22115856
PubMed Abstract | CrossRef Full Text | Google Scholar
Bita, C. E., and Gerats, T. (2013). Plant tolerance to high temperature in a changing environment: scientific fundamentals and production of heat stress-tolerant crops. Front. Plant Sci. 4, 273. doi:10.3389/fpls.2013.00273
PubMed Abstract | CrossRef Full Text | Google Scholar
Boopathy, L. R. A., Jacob-Tomas, S., Alecki, C., and Vera, M. (2022). Mechanisms tailoring the expression of heat shock proteins to proteostasis challenges. J. Biol. Chem. 298 (5), 101796. doi:10.1016/j.jbc.2022.101796
PubMed Abstract | CrossRef Full Text | Google Scholar
Cao, D., Wang, J., Ju, Z., Liu, Q., Li, S., Tian, H., et al. (2016). Regulations on growth and development in tomato cotyledon, flower and fruit via destruction of miR396 with short tandem target mimic. Plant Sci. 247, 1–12. doi:10.1016/j.plantsci.2016.02.012
留言 (0)