Participants
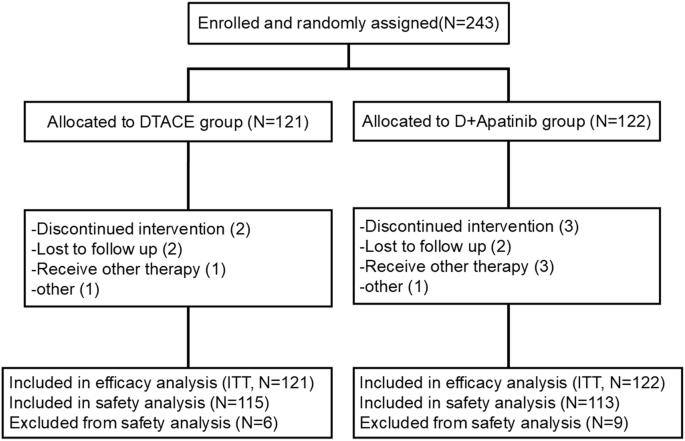
This phase III multicenter, randomized, open-label study was carried out in 12 hospitals in China from January 2021 to June 2022. Here, uHCC diagnosis was based on the “Diagnostic and Therapeutic Criteria for Primary Liver Cancer” guidelines in China,42 considering biopsy, cytology, or diagnostic imaging (dynamic computed tomography or magnetic resonance imaging). Participants with uHCC were randomly assigned 1:1 to the DEB-TACE alone (DEB-TACE) and DEB-TACE + apatinib groups through computerized central randomization using permuted blocks (sizes of four and six).
The key eligibility criteria were: 1) uHCC patients with recurrence/metastasis confirmed by histopathology or cytology, who strictly complied with the clinical diagnostic criteria of the “diagnostic and therapeutic criteria for primary liver cancer” (2017 Edition), were ineligible for palliative surgery or radiotherapy, and had ≥1 measurable lesion based on mRECIST criteria, requiring a long diameter ≥10 mm for the measurable lesion or a short diameter ≥15 mm for the enlarged lymph node); 2) after confirming the liver cancer, no treatments were performed before DEB-TACE, such as immunotherapy, liver transplantation, surgical resection, cTACE, radiofrequency/microwave/chemical ablation, argon helium knife, ultrasonic scalpel, radiation therapy, systemic chemotherapy, and targeted therapies, e.g., sorafenib, renfatinib, apatinib, and PD-1, programmed death ligand 1 (PD-L1), and cytotoxic T-lymphocyte associated protein 4 (CTLA-4) inhibitors; 3) BCLC stage B-C and non-diffuse liver cancer; 4) age of 18 to 75 years; 5) Eastern Cooperative Oncology Group (ECOG) performance status (PS) score 0-1 within 1 week pre-enrollment; 6) liver tumor accounting for <60% of total liver volume; 7) no serious complications, e.g., hypertension, coronary heart disease, and no history of mental disease or severe allergy; 8) liver function reaching Child Pugh grade A or B, and normal or post-treatment corrected renal and coagulation functions; 9) HBV DNA < 2000 IU/ml (104 copies/ml); 10) negative pregnancy test in females of childbearing potential within 7 days pre-enrollment; and 11) signing of the informed consent to participate in the trial and good compliance.
Exclusion criteria were: 1) imaging examination showing that the HCC liver tumor was huge ( ≥ 60% liver volume) or tumor thrombus in the main portal vein (occupying vessel diameter ≥50%), mesenteric vein or inferior vena cava invasion, or obvious arteriovenous fistula; 2) a history of liver transplantation, surgical resection, TACE, radiofrequency/microwave/chemical ablation, argon helium knife, ultrasonic scalpel, radiotherapy, or other local treatments, or systemic chemotherapy, oral targeted liver cancer drugs (sorafenib, renfatinib, or apatinib), or immunotherapy such as PD-1/PD-L1/CTLA-4; 3) diffuse liver cancer, known cholangiocarcinoma, mixed cell carcinoma, or fibrolamellar cell carcinoma, detected previously ( < 5 years) or other concurrent uncured cancers, except for skin basal cell carcinoma and cervical carcinoma in situ that have been cured; 4) grade ≥II myocardial ischemia or myocardial infarction and poorly controlled arrhythmia (QTc interval ≥450 ms in males and ≥470 MS in females); 5) gastrointestinal bleeding within 6 months or clear gastrointestinal bleeding tendency, e.g., esophageal varices with bleeding risk, local active ulcer lesions, or fecal occult blood ≥2+ (gastroscopy required in case of fecal occult blood at 1 + ); 6) impaired coagulation function, with INR > 1.5 or prothrombin time (PT) > ULN + 4 s), displaying bleeding tendency or under thrombolytic or anticoagulant treatment; 7) central nervous system or brain metastasis, previous and current confirmed pulmonary fibrosis, interstitial pneumonia, radiation pneumonitis, drug-related pneumonia, severe lung function impairment, HIV infection, pregnant or breast-feeding women, or scheduled liver transplantation (except for patients with previous liver transplantation); 8) expected OS < 3 months; 9) creatinine clearance rate (CCr) < 2 mg/min (CCr<2 mg/min), or 10) due to various reasons, not completed treatment plan, and no follow up within 3 months.
DEB-TACE
All participants were administered standardized DEB-TACE at each participating institution. The tumor-supplying artery was typically detected via hepatic angiography following the Seldinger puncture technique and abdominal trunk arteriography. The tumor-feeding artery was accessed using microcatheters via super-selective catheterization. CalliSpheres (Jiangsu Hengrui Pharmaceutical, China) containing 40-60 mg of doxorubicin or epirubicin (100-300 or 300-500 μm) were slowly administered by injection into the tumor-supplying artery, following previously reported techniques.20,21 In case of incomplete embolization, 350-560 μm polyvinyl alcohol (PVA) particles (Hangzhou Alikang Pharmaceutical Technology, Zhejiang, China) or 300-500 μm microspheres (Jiangsu Hengrui Pharmaceutical) could be additionally utilized.
Interventional radiologists with ≥10 years of experience conducted all TACE sessions at the respective participating centers. Intravenous analgesia, combining dexmedetomidine and dezocine, was administered for 48 h from TACE initiation to manage soreness during the procedure. Following DEB-TACE, the participants received 3–5 days of liver protection and symptomatic treatments to manage embolism syndrome symptoms. DEB-TACE was discontinued in case of disease progression or a condition making DEB-TACE infeasible (e.g., tumor thrombus in the main artery trunk or vascular injury), or persistent liver dysfunction, e.g., Child-Pugh score ≥9 points lasting for over 4 weeks.
Systemic therapy
Apatinib (Jiangsu Hengrui Pharmaceutical, Co., Ltd., Shanghai, China) was orally administered to the participants in the DEB-TACE + apatinib group for the first time 3-5 days after DEB-TACE, initially at 500 mg once daily. Apatinib administration was discontinued 3 days prior to a subsequent TACE session. In cases where participants experienced grade ≥3 AEs, the apatinib dose was reduced to 250 mg once daily, suspended, or discontinued. The maximal suspension period of apatinib was 2 weeks, with no more than two suspensions allowed. If symptomatic treatment failed to alleviate the observed AEs, discontinuation of apatinib was considered. The participants tolerating AEs at 250 or 500 mg once daily continued apatinib until tumor progression, intolerance, or death.
Assessment of treatment efficacy
Following the first TACE procedure, participants underwent routine blood analysis, liver and kidney function assessments, coagulation function evaluation, and tumor marker detection, along with enhanced magnetic resonance imaging (MRI) and/or computed tomography (CT) scans 4-6 weeks later. Two radiologists with extended experience evaluated the scans using mRECIST criteria to determine the curative effects (best response), i.e., complete response (CR), partial response (PR), stable disease (SD), or progressive disease (PD). The evaluation based on RECIST 1.1 was used for sensitivity analysis. ORR and DCR were obtained as CR + PR and CR + PR + SD, respectively. PFS was calculated from the date of first TACE to disease progression or death, and OS from the first TACE to death or last follow-up. Subsequent TACE procedures were performed if the tumor maintained arterial blood supply per enhanced MRI and/or CT with confirmed Child-Pugh classification A/B. Treatment was continued until untreatable progression, defined by meeting DEB-TACE refractoriness criteria, intolerable toxicity or consent withdrawal. During the embolization process of DEB-TACE, embolization with drug-loaded microspheres can create new spaces within the tumor mass, resulting in the accumulation of contrast agents. This imaging manifestation appears as an early arterial phase with slow disappearance, and the contrast agent can still accumulate until the venous phase, resembling a vascular lake.
Follow-up and safety evaluations
Complications associated with DEB-TACE included fever, nausea, vomiting, abdominal pain, constipation, ascites, liver abscesses, and hepatic artery thinning and spasm during the second DEB-TACE procedure. Apatinib-related AEs, e.g., hypertension, hand-foot syndrome, fatigue, diarrhea, gastrointestinal reactions, and liver dysfunction, were managed through symptomatic treatment. The examined participants or their families were queried about AEs, survival status, and cause of death (if indicated) post-treatment through outpatient visits, WeChat, and/or mobile phone communications. AEs were documented using the National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.03 (NCI-CTCAE 4.03).
After three or more consecutive standardized and refined TACE treatments, if enhanced CT/MRI conducted 1-3 months following the last procedure showed that the intrahepatic target lesion was still in a state of disease progression (PD) compared to before the first TACE treatment based on mRECIST criteria, TACE resistance was considered. In such cases, it was necessary to promptly preclude further TACE and switch to other treatments.
Sample size estimation
The primary endpoint was PFS. Based on previous reports and clinical experiences,40,44,45 the estimated median PFS for DEB-TACE + apatinib was approximately 9 months, versus around 6 months for DEB-TACE. The statistical parameters set for the test were: two-sided Class I error probability (α), 0.05; beta (β), 0.2; power, 0.8. The study aimed for a 1:1 ratio in both treatment groups. The PASS software calculated that at least 117 and 116 participants in the DEB-TACE + apatinib and DEB-TACE groups were required, respectively, considering a 5-15% potential loss to follow-up. Therefore, the total sample size for both treatment groups was 233 participants.
Statistical methods
Continuous variates were presented as mean ± standard deviation and analyzed by Student’s t-test. Categorical variates were analyzed by the chi-squared or Fisher’s exact test for comparison. Liver and kidney functions were analyzed using repeated measure analysis of variance. Kaplan-Meier curves were utilized to analyze survival outcomes, with differences assessed by the log-rank test. Factors influencing OS and PFS were evaluated through univariable and multivariable Cox proportional hazard regression analyses, and outcomes were expressed as hazard ratios with corresponding confidence intervals. The center effect was considered random due to the enrollment condition of this study (a total of 12 centers with some having a limited number of participants). Given the data type (survival data) and the need to adjust for random effects, a Cox Frailty model was employed to adjust for the center effect, and enrollment center was included as a random intercept.46

留言 (0)