
記住我
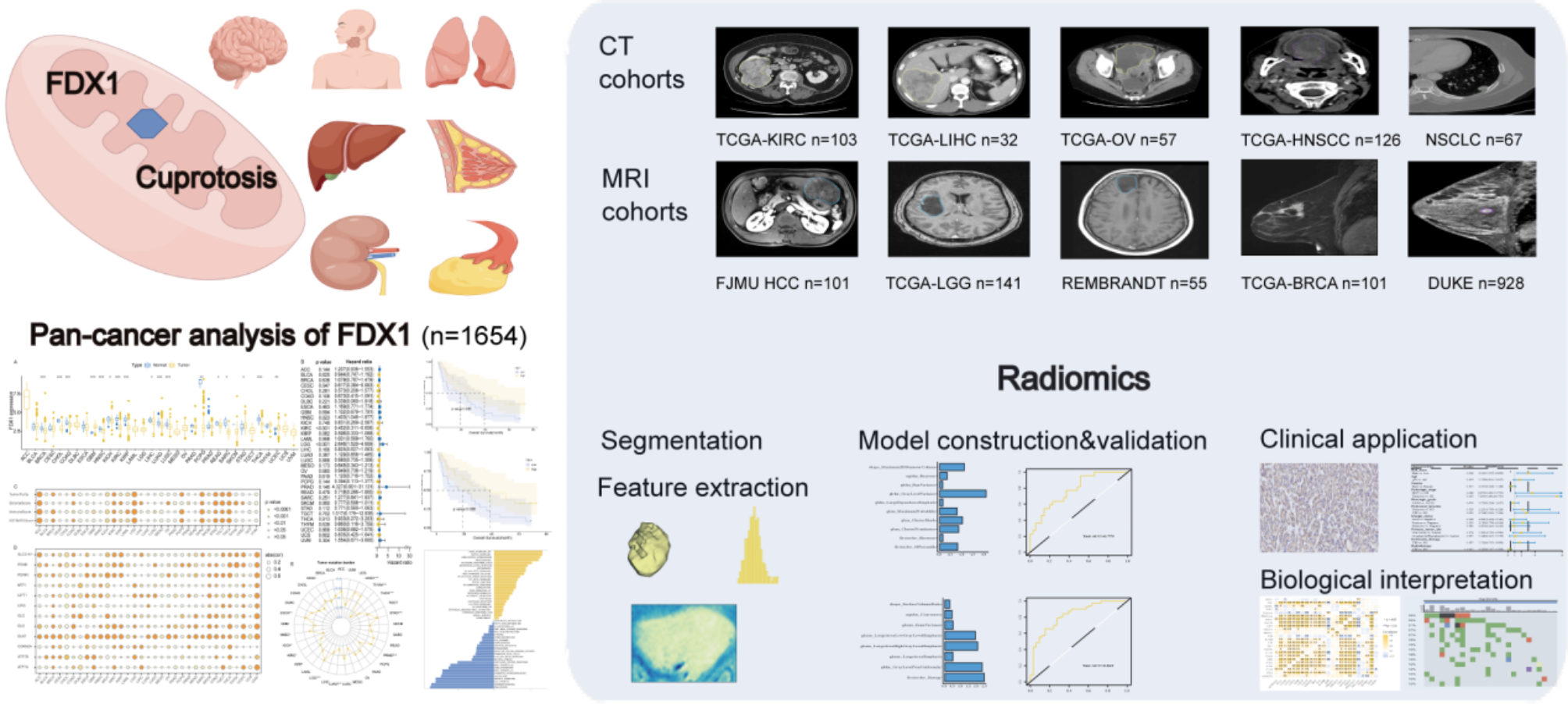

The flow of this study was depicted in Fig. 1. Transcriptome RNA sequencing data were downloaded: data on 33 cancer types from the Cancer Genome Atlas (TCGA), (https://tcga-data.nci.nih.gov/docs/publications/tcga/), non-small cell lung cancer (NSCLC) from Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE103584), and brain lower grade glioma (LGG) from the Chinese Glioma Genome Atlas (http://www.cgga.org.cn.portal.phpg); corresponding CT or MRI images were retrieved from the Cancer Imaging Archive, Gene Expression Omnibus, and REMBRANDT (http://caintegrator-info.nci.nih.gov/rembrandt). Duke breast cancer MRI images (https://sites.duke.edu/mazurowski/resources/breast-cancer-mri-dataset/) and clinical variables were retrieved as an independent test dataset for the prognostic value of the radiomic score.
For pan-cancer analysis, RNA sequencing data from TCGA, covering 33 types of cancer, were exploited. Corresponding clinical variables and survival outcomes were retrieved from TCGA to investigate the association between FDX1 expression and overall survival (OS) across cancer types.
The inclusion criteria were as follows: (1) primary tumors, (2) complete clinical data and (3) overall survival time over 30 days. The exclusion criteria were as follows: (1) bilateral lesions and (2) cases with unknown race, histologic grade, pathologic stage.
The inclusion and exclusion criteria for the public imaging cohorts were based on the previous studies [8]. The inclusion criteria were as follows: (1) solid tumors, (2) available important clinical data, (3) survive over 30 days. The exclusion criteria were as follows:1) non-primary tumors, 2) bilateral lesions, 3) no venous phrase images for CT cohorts or contrast-enhanced images for MRI cohorts, (4) poor image quality, and (5) postoperative imaging.
Fig. 1
The expression of FDX1 in different cell types was explored using single-cell sequencing datasets from the scTIME portal website [9]. Mutation annotation format files deposited in the TCGA data portal for somatic mutations were downloaded for analysis (https://portal.gdc.cancer.gov/).
The data of the HCC cohort diagnosed at the First Affiliated Hospital of Fujian Medical University between January 2017 and December 2020 was used to develop the MRI model. The inclusion criteria were (1) contrast-enhanced MRI images (3.0T) within 1 month before surgery; (2) complete pathological, imaging, and clinical data; and (3) good image quality. The exclusion criteria were (1) MRI images suggesting that the lesions invaded blood vessels, bile duct tumor thrombus, or extrahepatic metastasis and (2) a history of partial hepatectomy or interventional therapy. In patients with multiple lesions, the largest lesion was the preferred study object.
PreprocessingThe images were resampled to 1 × 1 × 1 mm3 pixels to eliminate the interference of inconsistent spatial resolution among the various models of imaging procedures.
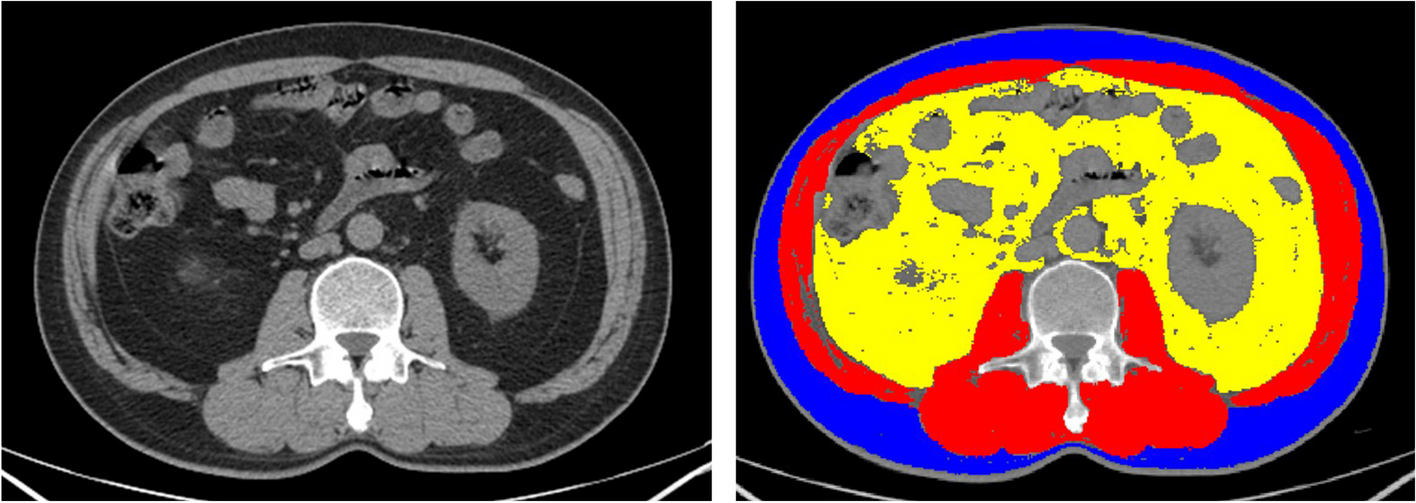
VOIs segmentationVOIs were outlined along the tumor contour by a radiologist (YQY, with > 9 years of experience) under double-blind conditions and verified by another radiologist (YRP, with > 6 years of experience in radiology). The entire tumor region was drawn manually using 3D Slicer software (https://download.slicer.org). During the segmentation, we made a comprehensive evaluation combined with other sequence/phase images, determined the lesion location by adjusting the appropriate window width and window position, and checked whether there were abnormal tissue masses, structural asymmetry, different density or signal, and abnormal enhancement areas to determine the tumor area.
Feature extractionWe extracted radiomic features using PyRadiomics (https://pyradiomics.readthedocs.io/en/latest/), and Z-score normalization was performed. It allowed us to derive both first-order statistics and higher-order texture features from the tumor volumes. Specifically, the extracted features included: 1) first-order statistics, which capture basic intensity values, such as mean, variance, skewness, and kurtosis; 2)shape features, which describe the geometry and size of the tumor, including metrics such as volume, surface area, and compactness; 3) texture features, derived from Gray-Level Co-occurrence Matrix (GLCM), Gray-Level Run Length Matrix (GLRLM), and Gray-Level Size Zone Matrix (GLSZM), which reflect spatial patterns and the heterogeneity of pixel intensities within the tumor.
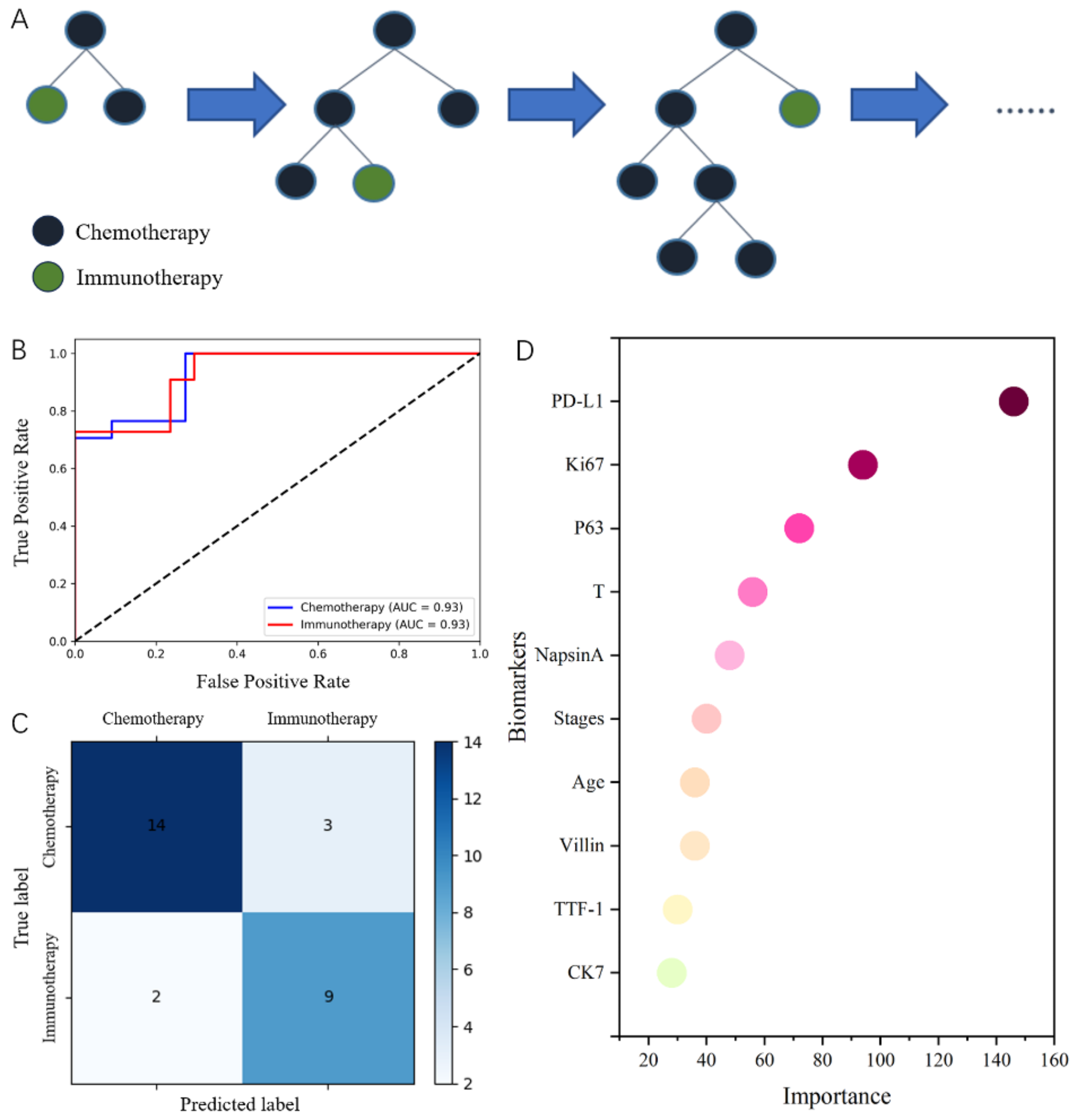
CT radiomic model for predicting FDX1 expressionGiven that the borders of the tumor in the venous phase were more distinct, features were screened on contrast enhanced KIRC CT images during the venous phase. To address the high dimensionality of the extracted features, we employed the Recursive Feature Elimination (RFE) algorithm to select the most relevant features. RFE iteratively ranks the features based on their importance in the model and removes the least significant ones. After this selection process, we reduced the number of features to 8 for the CT-based model and 13 for the MR-based model, retaining only the most relevant features for predicting FDX1 expression.
After feature selection, the remaining features were used to build the radiomic prediction model using logistic regression (LR). The selected features were input into the LR algorithm to model the probability of high vs. low FDX1 expression. This is a linear classification model where the log odds of the binary outcome are modeled as a linear combination of the selected radiomic features.
The probability of FDX1 expression predicted by the radiomic model was designated as the rad_score. To evaluate the radiomic model, ROC and precision-recall (PR) curves were plotted. The AUC and other diagnostic indices, including the BS, ACC, sensitivity, specificity, PPV, and NPV, were used to evaluate diagnostic performance. Calibration of the predictive model was demonstrated with calibration curves, and the Hosmer–Lemeshow test was used to assess the model fitness. Clinical usefulness was assessed using net benefits at different threshold probabilities determined by DCA.
We mitigated the risk of overfitting by applying RFE to select the most predictive features and ensuring a clear separation between training and test datasets. This allowed the model to generalize well to unseen data. In addition, we carefully split the datasets to ensure that the model was trained on one cohort and then validated and tested on independent cohorts from different cancer types. This approach ensured that the model was not overfitted to the training data and could generalize well across different cancer types and imaging modalities. By separating the training, validation, and test datasets, we minimized the risk of overfitting and provided a robust evaluation of the model’s performance on unseen data.
The kidney renal clear cell carcinoma (KIRC) cohort from TCGA served as the training dataset. The model was then validated using the liver hepatocellular carcinoma (LIHC) cohort from TCGA. The purpose of the validation was to fine-tune the model and evaluate its performance on an independent dataset from a different cancer type. To assess the generalizability of the model, it was tested on additional external cohorts, including the OV (ovarian cancer), HNSCC (head and neck squamous cell carcinoma), and NSCLC cohorts from TCGA. These test datasets provided a robust evaluation of the model’s performance across different cancer types and ensured that the model could generalize well to unseen data.
MR radiomic model for predicting FDX1 expressionThe MRI procedure for the hospital HCC cohort was as follows: T1C sequence after contrast agent injection was used for analysis, which had clearer comprehension of the anatomy. All MRI images were corrected using the N4 bias field correction algorithm, a popular method for correcting low-frequency intensity non-uniformity present in MRI image data, known as bias. The model was constructed using AP T1C images of the hospital HCC cohort. Then, the RFE algorithm was used to screen features, LR algorithm was used to model features, and rad_score was constructed to predict the expression level of FDX1. The model evaluation index was the same as that of the CT model. The model was verified using the TCGA-LGG cohort to check its performance on an independent dataset from a different cancer type. Finally, the model was tested on the REMBRANDT cohort (glioma dataset) and the Duke breast cancer cohort to evaluate its ability to generalize across additional cancer types and imaging modalities.
According to the exclusion criteria (poor image quality and postoperative imaging), 52 OV patients, 60 HNSCC patients, 104 NSCLC patients, 36 LIHC patients, 11 REMBRANDT patients, 29 BRCA patients, zero Duke patients were eliminated from patients with images.
IHCHCC tissues from the First Affiliated Hospital of Fujian Medical University were fixed, embedded in paraffin, and sectioned for IHC staining by an FDX1 antibody (Abmart, Shanghai, China). IHC was performed with antibodies targeting FDX1 (dilution 1:100), determined based on preliminary optimization experiments to ensure specific and robust staining. The stained tissue sections were counterstained with hematoxylin. Secondary biotinylated antibodies specific to the species of the primary antibody were used, followed by a horseradish peroxidase (HRP)-conjugated streptavidin for signal amplification.
Staining Protocol was as follows: Formalin-fixed, paraffin-embedded (FFPE) tissue sections from the HCC cohort were used for IHC. Tissue sections were cut at 4 μm thickness and mounted on glass slides. The tissue sections were deparaffinized using xylene and rehydrated through graded alcohols.
Antigen retrieval was performed using a citrate buffer (pH 6.0) at 95 °C for 20 min to enhance the accessibility of the FDX1 epitopes. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide for 10 min at room temperature. Tissue sections were incubated with the FDX1 primary antibody (1:100 dilution) overnight at 4 °C. The sections were then incubated with a species-specific biotinylated secondary antibody for 30 min at room temperature, followed by the HRP-conjugated streptavidin for 15 min. The signal was developed using a diaminobenzidine (DAB) substrate, resulting in a brown color for positive staining. The sections were counterstained with hematoxylin for contrast.
AIPATHWELL (Wuhan Servicebio Technology Co.), serves as a sophisticated tool for the comprehensive panoramic assessment of FDX1 IHC stained Sect. [10]. H-Score were calculated as (percentage of weak intensity*1) +(percentage of moderate intensity*2) +(percentage of strong intensity*3) [11]. Then H-Scores were categorized as low or high for further statistical analysis, based on the median cut-off.
Quantification and statistical analysisNormally and non-normally distributed quantitative data are presented as mean ± standard deviation and median (interquartile range), respectively, whereas categorical data are shown as percentages. The Wilcoxon rank-sum test was used to compare differences in quantitative data between the groups, whereas the chi-square and Fisher exact tests were used to compare differences in categorical variables between the groups.
All qualified cases were divided into two groups according to high and low FDX1 expression levels separated by the median value. Spearman or Pearson correlation coefficients were calculated using correlation analysis. Survival analysis was performed using Kaplan-Meier curves to compare the OS between high and low expression groups, and the log-rank test was used for significance testing. Univariate and multivariate Cox regression were applied to identify the hazard ratio (HR) of FDX1 expression in each cancer type. The prognostic significance of FDX1 was visualized through forest plots, depicting the hazard ratios. To identify differentially enriched pathways between FDX1high and FDX1low groups, gene set variation analysis was employed to calculate enrichment scores for the 50 hallmark pathways from the molecular signature database (MSigDB version 6.0).
Both immune and stromal scores were calculated using R packages “limma” and “estimate.” The correlation of FDX1 expression level with TMB was conducted via the R package “fmsb.” The rad_score was calculated, and patients were dichotomized by the R package “survMisc” for survival and correlation analysis. To compare the mutation differences between the high- and low-rad_score groups, visualizations of the highest frequency of mutations were performed in R using the maftools package. Each point on the x-axis of the waterfall plot represents an individual patient, used to calculate mutation frequency and quickly identify genes with high mutation frequencies in the sample.
The statistical significance level was set at a P-value (two-tailed) of < 0.05, except when specifically stated.
留言 (0)