This is a single arm, non-randomised, multicentre prospective study. In recognition of the strong international data that show benefit for PGx-guided dosing, it is considered unethical for us to conduct a randomised controlled trial to directly compare the implementation and non- implementation of PGx-guided dosing. It is a collaborative, investigator-led initiative supported by a trial committee comprising researchers from most states in Australia.
Study population
We aim to enrol 5000 patients from hospitals across multiple states in Australia and including metropolitan, regional and rural cancer services. Eligible participants are 18 years and older with solid organ malignancies intended to receive FP chemotherapies (either 5-FU or capecitabine) and/or irinotecan chemotherapy as part of either curative or palliative cancer management either as single agents or in combination. Patients must be able to provide informed consent and capable of providing a blood sample for genotyping. Exclusion criteria include those who have received prior treatment with FP chemotherapies, or who decline consent or blood collection. Patients who are pregnant or breastfeeding are also excluded, as are those already enrolled in other clinical trials that are likely to influence toxicity outcomes.
Intervention
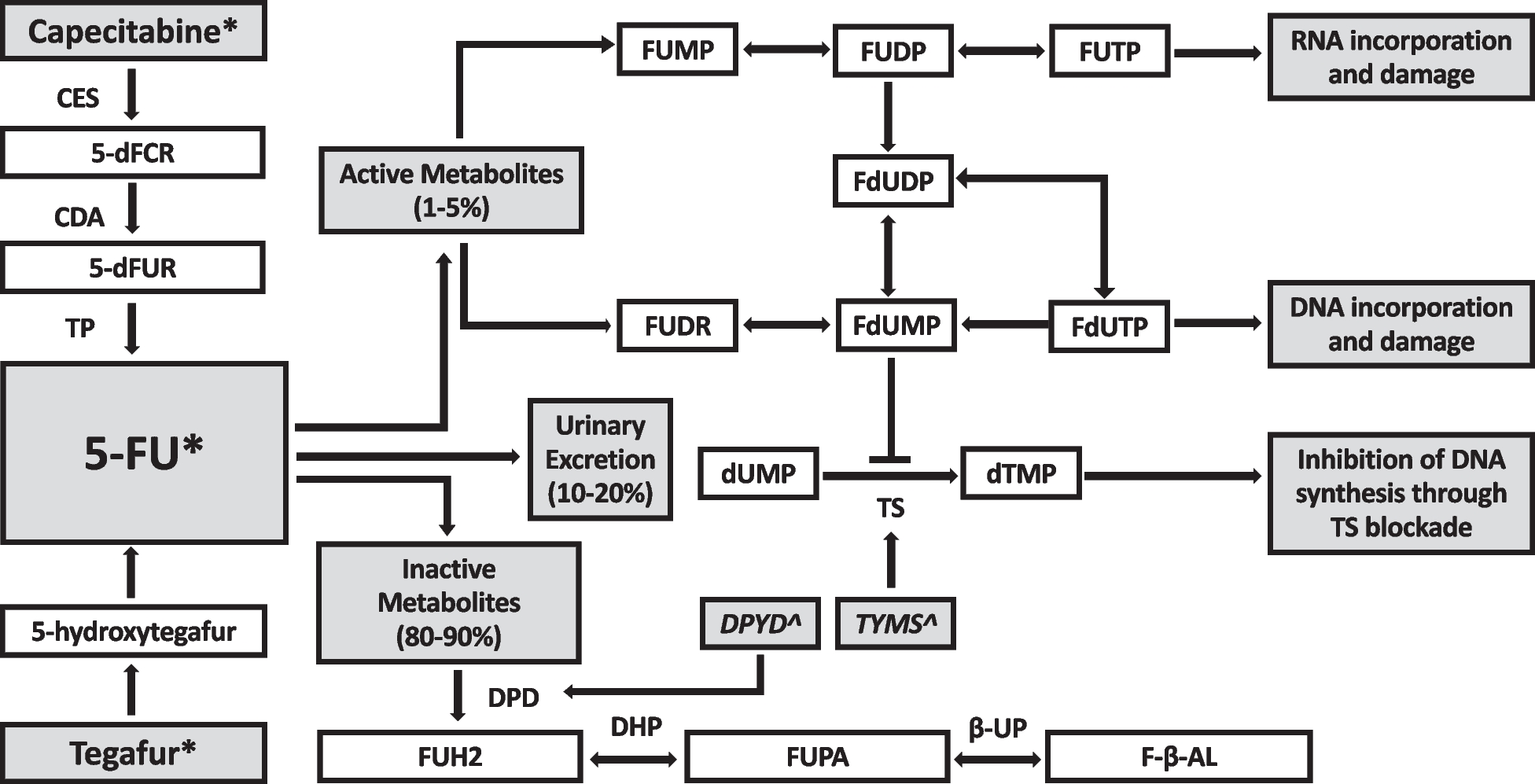
DPYD genotyping will be prospectively conducted in all patients prior to commencement of FP chemotherapies. Genotyping pathology services will be pre-determined by participating sites. Clinically significant DPYD variants will include c.1905 + 1G > A (rs3918290), c.2846A > T (rs67376798), c.1679 T > G (rs55886062) and c.1236G > A (rs56038477) (Supp Fig. 1). Patients found to carry one or more clinically significant variants will undergo dose-adjustment of FP chemotherapy prior to first administration, in accordance with eviQ guidelines (Supp Tables 1 and 2) [23].
◦ Heterozygote carriers of a single specified DPYD variant should receive a 50% dose-reduction prior to first FP exposure
◦ Following this administration, patients should be reassessed and FP dose further down-titrated or ceased in response to G3-4 toxicity, or up-titrated for those who experienced minor or no toxicities. Suggested increment for titration is ± 12.5% (of the 100% recommended dose), as suggested by EviQ.
◦ Patients who carry two variants (either compound heterozygote or homozygote carriers) should be dosed on an individual basis and may need to avoid FP administration altogether.
Data will be collected on all patients and will include specific datapoints for DPYD variant carriers who undergo dose-reductions to explore adherence to guidelines versus alternative dose-adjustment decisions. Bio-banked specimens from patients who do not have a pre-specified DPYD variant but experience ≥ G3 toxicities will undergo further exploratory genotyping. A subset of samples from patients intended to receive irinotecan chemotherapy will be batched and genotyped for clinically significant UGT1A1 variants (*6 (UGT1A1 c.211G > A), *28 A(TA)6TAA > A(TA)7TAA promoter), *37 (A(TA)6TAA > A(TA)8TAA promoter). Any UGT1A1 genotype-guided dose-adjustment will be at clinician discretion. UGT1A1 genotyping is for assessment of feasibility only. Data monitoring will be intermittently conducted by the trial management committee.
Implementation sub-study intervention
Qualitative interviews will be conducted with a convenience sample of 20 to 40 patients and clinicians involved in PGx screening, with the sample size dependent on achieving sufficient breadth and depth as is favoured over saturation in this methodology [29]. Interviews with patients will be conducted following enrolment, genotyping and at least first cycle of chemotherapy. Clinicians will be contacted throughout the trial. This will be decided by the research team through discussions parallel to recruitment and data collection. Participants will be asked to complete semi-structured qualitative interviews over the telephone or teleconferencing which will be up to 45 min in length. Patient interviews will explore the degree to which people offered genotyping were aware of and understood the purpose of genetic testing and dose-personalisation in the trial, experiences of DPYD variant carriers who received DPYD genotype-guided dose-personalisation, including information needs around, and lived-experiences of dose-personalisation throughout treatment. Clinician interviews will explore the experiences of using DPYD PGx-guided dose-personalisation, including reasons for adjusting/not adjusting the dose, intention to continue using DPYD PGx-guided dose-personalisation, and barriers and facilitators to this prescribing method.
Interview recordings will be transcribed, deidentified and imported into a qualitative data analysis software package for analysis. Interview and focus group transcripts will be deidentified prior to analysis. Only members of the research team who are not involved in patient care will be involved in the analysis. Reflexive thematic analysis will be conducted to explore the commonalities and dissimilarities within and across the data, and reflexive practice will be employed to acknowledge and account for any subjectivities (bias) in the data analysis [30]. These findings will be used to optimise the PGx screening process and provide tailored support and educational resources.
Clinical assessment, data collection and storage
The clinical assessment schedule is summarised below (Table 3), where implementation is included in the assessment table and is discussed separately below.
Table 3 GeneScreen 5-FU assessment scheduleInitial assessment will include patient demographics, tumour data and chemotherapy indication (curative/palliative). Chemotherapy regimens and FP dosing will also be collected. Clinical parameters including height and weight and haematological results prior to each cycle will be recorded. Dates of DPYD sample collection and results will be recorded, as well as the variant carrier status (including UGT1A1 where applicable).
Data from patients will be collected from clinical presentations within the first 60 days of their first FP dose to identify grade 3 and 4 toxicities requiring hospital presentation with or without admission, ICU admissions and deaths related to FP toxicities. Grading will be in accordance with the current CTCAEv5 [5]. For patients who carry DPYD variants and undergo dose-adjustments, FP dosing trends and adherence to/utility of the guidelines will be recorded. Patient follow-up data will be collected for 5 years to determine long-term cancer outcomes (DFS, PFS and OS). Data will be collected, de-identified and stored on a secure REDCap database. Patient samples will be de-identified and bio-banked.
Historical comparators
We felt it was unethical to have a control group of patients with DPYD variants who undergo standard FP dose administration due to the well described increased risk of toxicity within this population [9, 10]. Therefore, comparator populations for this project will include:
1)
Current standard of care, as assessed by a retrospective review of 500 consecutive cases in NSW [31].
2)
Contemporaneous cases without these DPYD variants treated with standard doses,
3)
DPYD variant carriers in this study that do not receive PGx-guided dose-adjustment, and.
4)
Patients treated in European cohorts whereby DPYD variant carriers were treated with a 25% or 50% FP dose reduction.
Background ≥ G3 toxicity in an ungenotyped population is approximately 17.4%, and in DPYD variant carriers without FP dose adjustment is reported to be 39–61% [8, 31]. Sub-analyses for each DPYD variant will be included.
Statistical considerationsStatistical hypotheses
We hypothesise that FP-induced severe toxicity (≥ G3) will decrease from 60% in the variant carriers receiving full dose chemotherapy to 35% in variant carriers receiving PGx-guided chemotherapy.
Sample size determination
This project intends to capture and test most feasibly large cohort of patients based on average clinical volume and anticipated number of patients that are eligible for testing across all study sites. Approximately 17000 patients are treated with FP per annum, resulting in > 30,000 potentially eligible patients across just 2 years of recruitment. As such, 5000 subjects in this study would require a recruitment capacity of < 15%. Our participating sites provide care for > 30% of Australians with cancer.
Based on expected frequency of DPYD variant alleles (~ 4% as a conservative estimate), testing N = 5000 patients will result in ~ 200 patients with actionable genotypes. Toxicity in these patients will be compared to historical datasets (Table 4). Assuming toxicity decreases to 35% in those receiving PGx- guided dosing, N = 70/arm will provide 80% power to detect a significant improvement in FP-related toxicity at the 5% level.
Table 4 Populations for analysisStatistical analyses
Descriptive statistics on continuous data will include means, medians, standard, deviations, and ranges, while categorical data will be summarized using frequency, counts and percentages. Graphical summaries of the data may also be presented.
The 95% confidence intervals for the proportion of tests returned within 7 days of intended chemotherapy dosing and the proportion of PGx guided dosing adjustments implemented will be estimated using the Clopper-Pearson method. PGx-guided dosing will be deemed feasible to implement if both 95% confidence intervals are completely above 80%.
The proportion of DPYD positive patients experiencing ≥ G3 toxicities in the current sample will be compared to the historical controls using a Fisher’s exact test. Logistic regression will also be used to compare the rate of ≥ G3 toxicities between the two groups. The intervention effect will therefore be summarised using the odds ratio and 95% CI from this model.
Health economic analysis
A modelled cost-effectiveness analysis will estimate the incremental cost per QALY gained of DPYD screening. Prevalence, intervention costs and toxicity-related hospital costs (associated with inpatient separations, emergency department presentations and outpatient care) incurred during the study period will be used to populate a decision analytic model with a one-year time horizon. Health outcomes and hospital costs for a comparator group (i.e. no genotyping) will be estimated using published literature, analysis of a linked dataset (from historical cohorts) and prospectively in GeneScreen 5-FU participants that have a known DPYD genotype but did not have dose-adjustment. One-way sensitivity analyses will be conducted for key model parameters including DPYD screening costs, prevalence of DPYD genotypes and health outcomes. Probabilistic sensitivity analyses will be conducted to evaluate the impact of parameter uncertainty on model outcomes and will be presented on a Cost-Effectiveness Acceptability Curve for a range of cost-effectiveness thresholds. A budget impact analysis will be developed to inform on the affordability of national uptake of DPYD testing to support implementation.
Implementation strategies
Semi-structured qualitative interviews of patients and clinicians will explore the attitudes and opinions to understand the experiences of those involved in DPYD/UGT1A1 PGx screening. Forty representatives from patient and clinician groups will be selected at random. These data will be used to optimise the PGx screening process and provide tailored support and educational resources where required. Interviews with patients will be conducted following enrolment, genotyping and at least the first FP cycle. Clinicians will be contacted throughout the trial. Interviews will be recorded, de-identified and securely stored on password-protected university servers.
Translational research
A portion of patients will develop ≥ G3 toxicities without being carriers of one of the genotyped DPYD variants, suggesting they either carry different clinically significant DPYD variant(s) or other genetic/epigenetic drivers of FP toxicity yet to be identified. We will conduct additional gene sequencing on bio-banked specimens to determine other genetic influences on FP toxicity. Furthermore, we will be able to provide further data regarding the limitations of linkage disequilibrium of c.1236G > A and HapB3 haplotype (c.1129-5923C > G, rs75017182) as described by Turner et al. [32]. This information will ideally help to tailor a PGx panel specific to our diverse Australian population to assist with identifying maximum numbers of patients at risk of toxicity prior to first FP exposure.


留言 (0)