
記住我

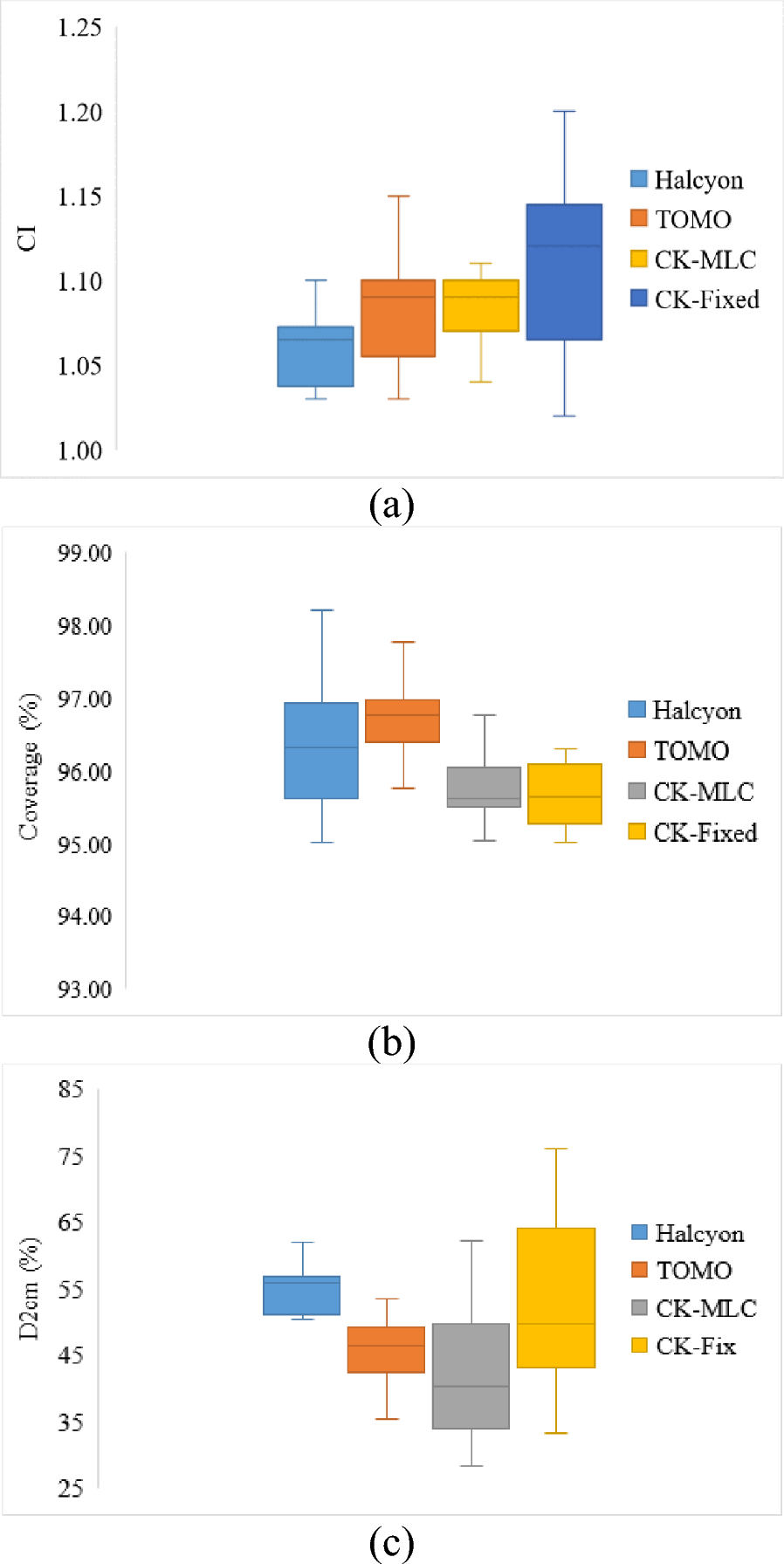
A total of 42,624 newborns were collected, with 23,143 samples obtained from Gansu Provincial Maternity and Child-care Hospital in southeastern China between 2017 and 2020, and 19,481 samples collected from Children’s Hospital Affiliated with Shandong University in eastern China from 2016 to 2021. All samples carrying information on other genetic metabolic disorders or treatment were excluded, comprising 51 MMA patients (46 from Shandong, 5 from Gansu) with treatment details and 569 patients (124 from Shandong, 445 from Gansu) diagnosed with various other metabolic disorders. The 51 excluded MMA patients included cases where MMA developed during the neonatal period and treatment began before screening. The 569 individuals excluded due to other metabolic disorders had received a definitive diagnosis through genetic testing. These disorders included conditions such as phenylketonuria, maple syrup urine disease, and homocystinuria, among others. The control group consisted of non-affected individuals who were matched for age, sex, and geographical location. These individuals had no known metabolic disorders and did not present with any screening abnormalities indicative of MMA or other metabolic conditions. Each MMA patient has a definite pathogenic mutation confirmed by Sanger or Next-generation sequencing. This study protocol was approved by the Ethics Committee of the National Research Institute for Family Planning (Beijing, China). The personal information for all newborn samples was deleted to safeguard each person’s privacy.
Metabolic analytes of the newborn screeningThe small molecular metabolite analytes were detected from the dried blood spot (DBS) samples by MS/MS. There are 45 features in total including 10 amino acids metabolic analytes, including Alanine (Ala), Glycine (Gly), Proline (Pro), Valine (Val), Methionine (Met), Phenylalanine (Phe), Tyrosine (Tyr), Citrulline (Cit), Ornithine (Orn), Arginine (Arg); 31 fatty acids metabolic analytes, including Free carnitine (C0), Acetyl-carnitine (C2), Propionyl-carnitine (C3), Dicarboxybutyl-carni tine (C5OH+C4DC), Butyryl-carnitine (C4), Dicarboxypropyl-carnitine (C4OH+C3DC), Octenoic-carnitine (C5), Isohexenoyl-carnitine (C5:1), Pentadecanedioyl-carnitine (C5DC+C6OH), Hexanoyl-carnitine (C6), Octenoic-carnitine (C8:1), Octanoyl-carnitine (C8), Decadienoic-carnitine (C10:2), Decenoic-carnitine (C10:1), Decanoyl-carnitine (C10), Dodecenoyl-carnitine (C12:1), Dodecanoyl-carnitine (C12), Tetradecadienoic-carnitine (C14:2), Tetradecenoic-carnitine (C14:1), Tetradecanoyl-carnitine (C14), Hydroxytetradecanoyl-carnitine (C14OH), Hexadecenoic-carnitine (C6:1), Hexadecanoyl-carnitine (C16), Hydroxyhexadecenoic-carnitine (C16:1OH), Hydroxyhexadecanoyl-carnitine (C16OH), Octadecadienoic-carnitine (C18:2), Octadecenoic-carnitine (C18:1), Octadecanoyl-carnitine (C18), Hydroxyoctadecenoic-carnitine (C18:1OH), Hexanedioyl-carnitine (C6DC), Hydroxyoctadecanoyl-carnitine (C18OH); and the ratios of 4 fatty acids, which are Propionyl-carnitine/Free carnitine (C3/C0), Propionyl-carnitine/Acetyl-carnitine(C3/C2), Propionyl-carnitine/Hexadecanoyl-carnitine (C3/C16), and Phenylalanine/Tyrosine (Phe/Tyr). Phe/Tyr is a common indicator in NBS, and C3/C2, C3/C0, and C3/C16 are closely related to MMA [16]. Descriptive statistical information for each of the 45 features utilized in this study is provided in Additional file 1: Table S1.
Genetic analysis of MMA patientsGenomic DNA preparationA total of 2–3 ml of blood samples were collected from the probands and their parents. Genomic DNA was extracted using the Tiangen Biotech DNA extraction kit (Beijing, China).
Whole-exome sequencing (WES)Whole exome sequencing was performed using an Agilent SureSelect Human All Exon V6 Kit (Agilent Technologies Inc., USA) on an Illumina NovaSeq 6000 platform (Illumina Inc., CA, USA). This capture sequencing provides approximately 99% coverage of the target sequence, with an average depth > 20× coverage of 99%. Variants were described according to the nomenclature recommended by the Human Genome Variation Society (www.hgvs.org/). Variant frequencies were searched for in the GnomAD (http://gnomad.broadinstitute.org/), Exome Sequencing Project (ESP, http://evs.gs.washington.edu) and SNP (dbSNP) (http://www.ncbi.nlm.nih.gov/projects/snp) databases. Candidate variants were confirmed in the parents of each family by Sanger sequencing. Variants were checked in the Human Gene Variant Database (www.hgmd.cf.ac.uk) and ClinVar database (www.ncbi.nlm.nih.gov/clinvar/). InterVar (http://wintervar.wglab.org/) software was used to evaluate the pathogenicity of all variants according to the standards and guidelines of the American College of Medical Genetics and Genomics (ACMG) [17].
Data analysisThe results obtained from WES were compared with the reference genome (GRCh37/hg19), and the detected high-quality variants were annotated with variant information according to the PGenomics platform (https://pgenomics.cn), a national shared service platform for human genetic resources. What’s more, PGenomics platform can combine information on the clinical phenotype, inheritance pattern, family codisjunction, and pathogenicity of the various point of the affected children to screen the detected candidate variation point and score them comprehensively by bioinformatics software; the higher the score, the higher the correlation. All the candidate causing variants were checked manually.
Sanger sequencingCandidate variants were confirmed in the parents in each family by Sanger sequencing. PCR products were bi-directionally sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, USA) on an ABI 3500DX Genetic Analyzer (Applied Biosystems) after purification on 2% agarose gels.
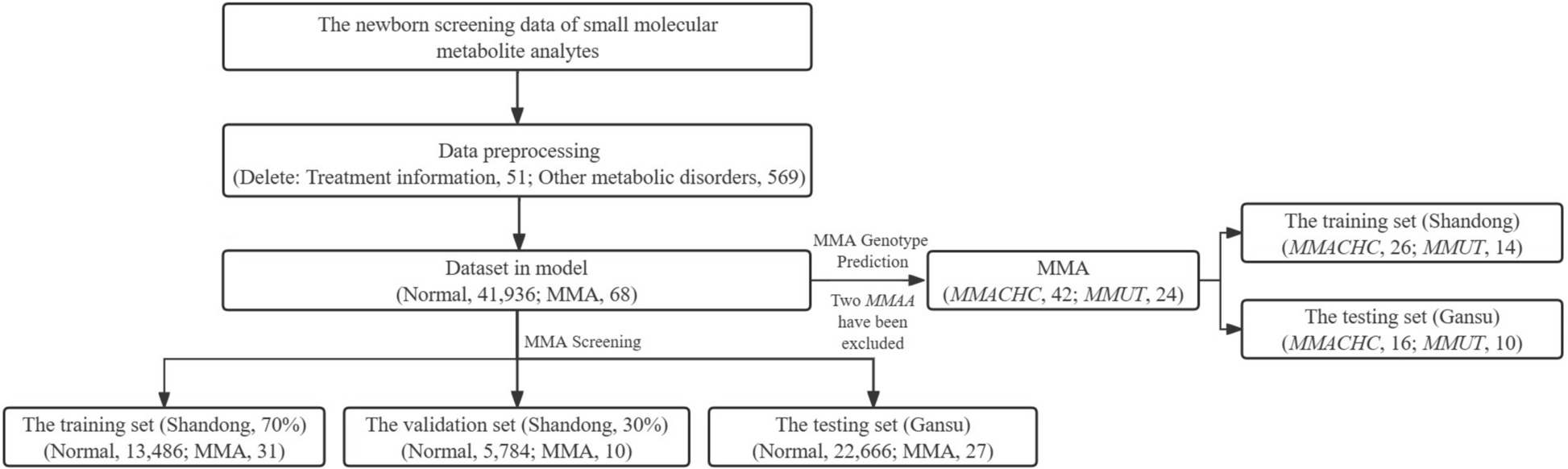
Metabolic data sets and data processingThe entire study population was stratified into two distinct categories: individuals diagnosed with MMA and healthy controls devoid of the condition. Within the assemblage of MMA subjects, three primary genotypes were identified: MMUT, MMACHC, and MMAA. Upon through completion of the specified preprocessing procedures, the data set originating from Shandong province, encompassing a cohort of 41 MMA patients (MMACHC, 26; MMUT, 14; and MMAA, 1) and 19,270 normal newborns, underwent a rigorous randomization process, with a 7:3 allocation ratio, to establish both training and validation sets in MMA screening. As a result, the training data set comprised 13,486 normal newborns and 31 MMA patients, while the validation set consisted of 5784 normal newborns juxtaposed against 10 MMA patients. Separately, an independent testing set was derived from Gansu Province, incorporating a total of 22,693 samples, including 27 MMA patients (MMACHC, 16; MMUT, 10; and MMAA, 1) and 22,666 normal newborns. Due to the scarcity of MMAA genotype data, the two patients with MMAA genotypes were excluded, and only 66 patients (Shandong, 40; Gansu, 26) carrying MMACHC and MMUT genotypes were selected for constructing the subdivision model of MMA genotype prediction. Following the training of various models on the training set, the performance of each model is measured and judged using the validation set. Therefore, the validation set can be used for model selection. The testing set is only used once after training to evaluate the generalization of the model. Figure 1 depicts the general process of data analysis in MMA screening and genotype prediction, offering a comprehensive overview of the sequential stages involved, from initial data acquisition to the final evaluation stage.
Fig. 1
General process for building an MMA screening and MMA genotype prediction model on MS/MS data
Models trainingSeven machine learning models were trained to separate MMA patients from normal newborns, including Linear Regression (LR), Support Vector Machine (SVM), Decision Tree (DT), Stochastic Gradient Descent (SGD), Multi-Layer Perceptron (MLP), and RFC. The 45 feature variables were input into the machine learning models as continuous variables. For all machine learning models, these 45 features were directly input into the models without undergoing any further transformation. All models were optimal models after parameter adjusting and were computed using Scikit-learn 1.0.1 in Python 3.8.5.
Random forest classifierRFC is a combined classifier algorithm proposed by Breiman in 2001 [18]. It is a supervised learning algorithm and an ensemble learning algorithm based on the decision tree. The RFC constructs an ensemble of k trees, each trained on a bootstrapped data set subset via Bagging. At each node split, a random subset of features is considered to enhance diversity. Trees are grown to maximize node purity without pruning. Prediction involves aggregating outputs from all k trees for improved decision-making.
To achieve the optimal RFC model, the number of trees in the forest, a maximum depth, a minimum split size, and a minimum leaf sample size were fine-tuned by the Python library’s “Grid Search” in this study. Due to the imbalance between MMA and non-MMA samples, we generated category weights with high weights for small number of samples and low weights for large number of samples. The ideal criterion for clinical practice in MMA screening is to simultaneously detect all MMA patients with excellent PPV. Each decision tree determines the MMA or non-MMA status of its own sample when one is added to the MMA screening model. By combining the disease status of each decision tree and employing a straightforward voting method with the minority following the majority, the model decides whether the sample is an MMA patient.
Similarly, each decision tree determines the MMACHC or MMUT status of its own sample when one is added to the MMA genotype prediction model. By combining the MMACHC or MMUT status of each decision tree and employing a straightforward voting method with the minority following the majority, the model decides whether the genotype of the MMA patient is MMACHC or MMUT.
Feature importanceGini impurity is the likelihood of incorrectly classifying a randomly chosen element in a data set based on its class distribution. The feature importance in RF represents the total reduction of Gini impurity on all nodes split based on the feature. The lower the Gini impurity, the higher the purity, the higher the order of the collection, and the better the classification effect.
Model performanceThis work uses RFC to solve a binary classification problem. To see the accurate and incorrect class of each MMA status of the sample, the confusion matrix is employed (Table 1).
Table 1 Confusion matrix in MMA screeningWe employed an imbalanced data set for MMA screening. Since there are significantly more non-patient records than there are MMA patient records, accuracy cannot be the only metric. Then we assessed the performance of the classification using accuracy, sensitivity, specificity, false positive rate (FPR), and positive predictive value (PPV), as shown in Eq. (1). In addition, the area under the receiver operating characteristic curve (AUC) was utilized to evaluate the performance of the model, and the area under the receiver operating characteristic (ROC) curve is called AUC:
$$} = \frac} + }}}} + } + } + }}},$$
$$} = \frac} + \text}},$$
$$} = \frac} + \text}},$$
(1)
The Pearson chi-square test is used to test whether two categorical variables are independent of each other. It is a hypothesis-testing technique based on the chi-square distribution.
留言 (0)