
記住我

Significant advancements were made in understanding the genetic underpinnings of neurodegenerative diseases (NDs), specifically Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), and spinocerebellar ataxia (SCA), facilitated by the application of WES. Through comprehensive analyses, WES has uncovered rare variants, novel mutations, and crucial insights into the genetic complexities associated with these debilitating conditions.
Alzheimer’s diseaseWES has played a pivotal role in advancing our understanding of AD genetics, particularly in early-onset cases. Contrary to the expected prevalence of causative variants in major AD genes, such as APP, PSEN1, and PSEN2, WES has revealed a lower frequency in both familial late-onset AD and sporadic cases [25]. Moreover, WES studies have highlighted the significance of ultra-rare, loss-of-function variants in the SORL1 gene, linking them to an earlier onset of AD [44]. Notably, analyses in both AD cases and controls have uncovered rare, damaging variants in genes associated with amyloid-β processing, lipid metabolism, and microglial function, providing insights into the multifaceted pathogenesis of AD [37]. Rare variants in known AD risk genes, such as AKAP9, CD33, and CR1, were also identified, pointing to links between AD, immunity, neuronal structure, and mitochondrial function. The discovery of genetic links across multiple families suggests potential significance in other neurological pathologies, such as Charcot–Marie–Tooth and other synapse dysfunctions [45].
It is important to distinguish between causative genes and risk factors in AD genetics. APP, PSEN1, and PSEN2 are causative genes, meaning mutations in these genes directly lead to the development of AD. On the other hand, genes such as SORL1, as well as others mentioned above, are risk factors associated with the disease. These risk factors are identified through genome-wide association studies (GWAS), which help find genetic similarities associated with specific diseases, allowing for a deeper understanding of their genetic architecture.
Parkinson’s diseaseIn the realm of PD, WES has elucidated novel mutations in the CSMD1 gene, a complement control protein associated with inflammation in the CNS and previously linked to PD risk [28]. These findings reinforce the potential of the complement pathway as a therapeutic target for PD. Among pure PD forms, VPS35 and VPS13C are two genes discovered by WES [46, 47]. Many others are identified in complex forms of PD (DNAJC6, and SYNJ1), while others are awaiting confirmation (CHCHD2, DNAJC13) [46, 47]. Additionally, WES has identified enrichments in genes related to the extracellular matrix and regions previously implicated in PD by GWAS [48]. Notably, the gene RAD51B, known for its protein interaction with RAD51, has been associated with congenital mirror movements and comorbidities with PD [48].
GWAS have also provided deeper insights into PD’s genetic architecture. These studies analyze the entire genome to identify variations associated with the disease. Notable genetic risk factors include SNCA, LRRK2, and MAPT, which influence protein aggregation and mitochondrial function. For example, SNCA variants affect alpha-synuclein protein aggregation, a fundamental trait of PD, while more common mutations such as LRRK2 affect kinase activity and neuronal health [49, 50].
Furthermore, additional genes such as NRXN2 have been implicated in hereditary PD. A study involving a South African family with autosomal dominant PD identified a possible pathogenic mutation in the NRXN2 gene using WES. The NRXN2 variant showed consistency regarding absence within unaffected family members and controls, and was expressed in the substantia nigra [51]. The TMEM protein family genes (TMEM230, TMEM59, TMEM108) have also shown potential associations with PD, through their role in the regulation of vesicular trafficking and autophagy [52]. However, subsequent studies in other populations, including Chinese and Caucasian cohorts, have shown mixed results regarding the prevalence and significance of TMEM230 mutations [53].
Furthermore, the application of WES has expanded our understanding of early-onset PD genetics in specific ethnic populations, revealing new homozygous pathogenic variants in the PRKN, PARK7, and PINK1 genes in Iranian patients [29]. Likewise, an investigation conducted among ethnic Chinese participants demonstrated that 7.5% exhibited pathogenic variants in established PD genes, yielding notable results in the identical genes observed in the Iranian cohort [54].
Other NDsWES has been instrumental in delineating the genetic landscape of ALS and SCA. For ALS, at least 24 genes have been identified as associated with the disease. Among these, KIF5, NEK1, and ATXN2 are notable risk factors. The identification of a UBQLN2 mutation, unique to ALS without frontotemporal dementia, and its association with key neuronal proteins in inclusions underscore the specificity of WES in unveiling disease-specific mutations [36]. A noteworthy revelation establishes a connection between mutations in the valosin-containing protein (VCP) gene and ALS, extending its previously known associations with other NDs like body myopathy, Paget’s disease, and frontotemporal dementia [55]. In the case of SCA, WES has revealed a critical mutation in the ITPR1 gene, linking it to both congenital non-progressive SCA and adult-onset SCA type 15 [56]. Additionally, WES has identified mutations in genes encoding voltage-gated potassium channels, highlighting the pivotal role of ion channels in regulating neuronal excitability and contributing to cerebellar degeneration [34].
Hereditary ataxias, including SCA, are a large group of neurodegenerative disorders which are defined by progressive cerebellar ataxia. These disorders are often caused by mutations in various genes responsible for maintaining cerebellar function and integrity. The identification of such mutations through WES has significantly advanced our understanding of the molecular mechanisms underlying these conditions.
Furthermore, WES has significantly improved diagnostic success. In a study of 76 diverse families with sporadic or familial cerebellar ataxia, excluding common SCAs and Friedreich ataxia, WES yielded definitive or probable diagnoses in 32% of the cases [57]. The most prevalent mutations that emerged were in the RFC1, KIF1A, and SYNE1 genes. This underscores the efficacy of WES in uncovering the genetic underpinnings of relatively rare cerebellar ataxia within a diverse cohort [57]. Other hereditary ataxias include, Ataxia–Telangiectasia (A–T), caused by mutations in the ATM gene. This gene is responsible for DNA repair, meaning mutations often lead to progressive cerebellar degeneration, immunodeficiency, and an increased risk of cancer [58]. Although the ATM gene was identified prior to the advent of NGS, its large size made genetic diagnosis difficult; however, WES has improved the analysis of this gene, facilitating the molecular diagnosis of A–T. Finally, WES has advanced the analysis of causal genes for Wolfram syndrome (WFS). For example, recently, WES identified two novel homozygous variants in the WFS1 gene in Moroccan families: a missense mutation (c.1329C>G; p.Ser443Arg) and a nonsense mutation (c.1113G>A; p.Trp371Ter) [59]. These variants, which affected conserved amino acid residues and were absent from genetic databases and Moroccan controls, were validated as pathogenic through bioinformatics analysis and molecular modeling. This application of WES not only pinpointed the specific genetic causes of WFS in these families but also expanded the known mutational spectrum of the disease, demonstrating WES’s efficacy in diagnosing and understanding rare genetic disorders [59].
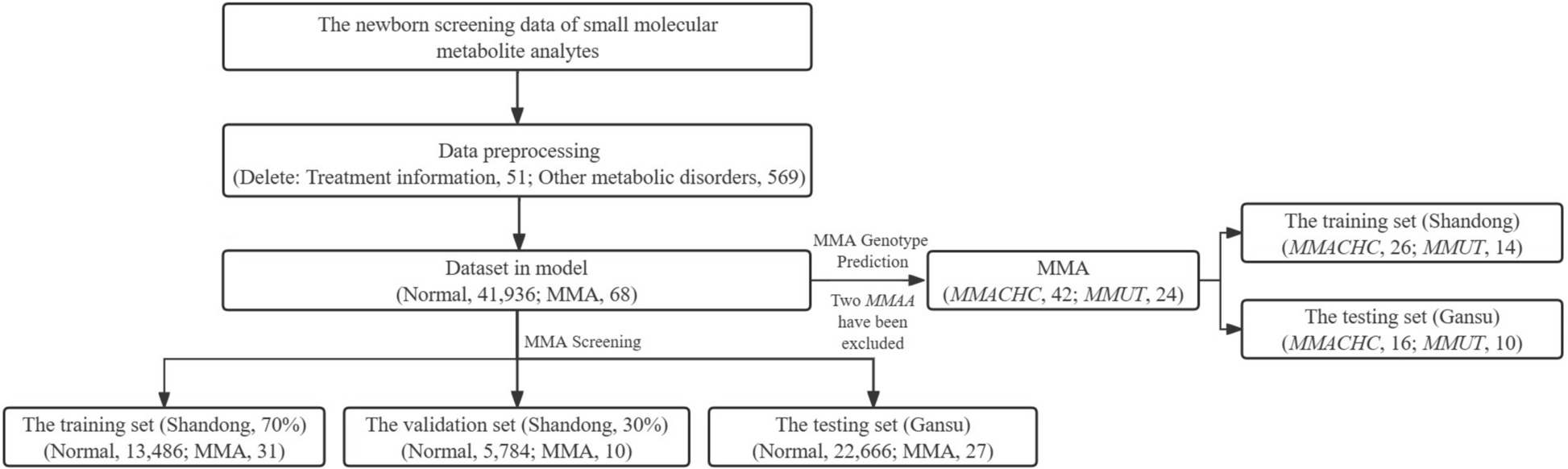
The collective findings stemming from WES applications in delineating the genetic landscapes of various NDs hold promise for therapeutic interventions. By uncovering rare variants, novel mutations, and disease-specific genetic signatures, WES offers avenues for developing targeted and personalized treatments for patients grappling with these complex conditions. The role of WES in NDs has been illustrated in Fig. 1.
Fig. 1
Role of whole-exome sequencing in neurodegenerative diseases. AD, Alzheimer’s Disease; ALS2, Alsin Rho Guanine Nucleotide Exchange Factor ALS2; APP, Amyloid Precursor Protein; C9orf72, Chromosome 9 Open Reading Frame 72; CHMP2B, Charged Multivesicular Body Protein 2B; CSMD1, CUB and Sushi Multiple Domains 1; DCTN, Dynactin; NEFH, Neurofilament, Heavy Polypeptide; OPTN, Optineurin; PARK7, Parkinsonism-Associated Deglycase (also known as DJ-1); PD, Parkinson’s Disease; PFN1, Profilin 1; PINK1, PTEN-Induced Putative Kinase 1; PRKN, Parkin RBR E3 Ubiquitin Protein Ligase (also known as PARK2); PRPH, Peripherin; PSEN1, Presenilin 1; PSEN2, Presenilin 2; RAD51B, RAD51 Paralog B; SIGMAR1, Sigma Non-Opioid Intracellular Receptor 1; SOD1, Superoxide Dismutase 1; SPG11, Spastic Paraplegia 11 (autosomal recessive); SQSTM1, Sequestosome 1; TBK1, TANK-Binding Kinase 1; TUBA4A, Tubulin Alpha 4a; UBQLN2, Ubiquilin 2; VAPB, Vesicle-Associated Membrane Protein, Associated Protein B and C; VCP, Valosin-Containing Protein; WES, Whole-Exome Sequencing
Cerebrovascular diseasesWES emerges as a powerful tool for deciphering the complex genetic underpinnings of cerebrovascular diseases (CVDs), offering unprecedented insights into diagnostic challenges and unveiling potential therapeutic targets [60].
Intracerebral aneurysmsWES has proven instrumental in identifying a spectrum of novel risk genes associated with intracranial aneurysms (IAs), elucidating key genetic contributors to CVDs. Notable instances include the identification of EDIL3 and TMEM132B genes as potential risk genes through WES, with EDIL3 substantiated by functional assessments and significant overexpression of the TMEM132B gene observed in IA tissue, linking both genes to the development and rupture of IAs [61, 62]. In a distinct ethnic context, the discovery of a missense variant (c.2519T>C, p.Leu840Pro) in the NFX1 gene highlighted its specific association with a heightened prevalence of IAs in a Chinese family, underscoring the diverse genetic landscape of IA pathology [63]. Concurrently, potentially deleterious variants within the PLOD3, NTM, and CHST14 genes were unveiled by WES, establishing them as primary causative factors for Familial IAs (FIAs) in Korean families [64]. Additionally, through GWAS and WES, more than 20 IA-candidate loci have been identified as risk factors. These collective findings underscore the indispensable role of WES in unraveling the intricate genetic landscape associated with IA.
StrokeWES has played a pivotal role in advancing our comprehension of genetic factors linked to stroke, with a particular focus on ischemic stroke. Noteworthy genetic insights have been gleaned through the identification of mutations occurring in exon 11 of the TRPV3 gene [35]. Additionally, two novel genes, PDE4DIP and ACOT4, have been associated with an elevated risk of ischemic stroke, providing novel dimensions to our understanding of the genetic basis of this CVD [65]. Furthermore, the investigation of a rare variation within the PON enzyme gene has revealed its capability to alter enzyme function, thereby increasing the susceptibility to ischemic stroke, particularly among individuals of African ancestry [66].
Sneddon syndrome, a rare genetic disorder, manifests as ischemic strokes predominantly affecting young individuals, particularly females [30]. It has elucidated that Sneddon syndrome is usually caused by bi-allelic ADA2 gene pathogenic variants. Moreover, the impairment of NOTCH3 signaling is not associated with Sneddon syndrome but is the causative gene of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), a different genetic disorder characterized by recurrent strokes and dementia [30]. Moreover, the specificity of WES in dissecting genetic contributors to distinct CVD subtypes was underscored by the identification of noteworthy genetic variants. For instance, while deleterious variants in KRIT1 and NOTCH3 are well-known causes of intracerebral hemorrhage and strokes, respectively, the recognition of new KRIT1 variants and rare NOTCH3 variants, such as p.R544C for ischemic small vessel disease, highlights the precision of WES. This precision allows for the identification of previously unknown genetic variations associated with hemorrhagic and ischemic subtypes of CVDs [33]. These collective findings underscore the promising role of WES in identifying causative genes for specific subtypes of CVDs, offering prospects for personalized diagnostics and targeted therapeutic interventions.
Other CVDsWES has been pivotal in advancing our understanding of a myriad of other CVDs. In a study focusing on subarachnoid hemorrhage (SAH), WES has pinpointed 30 SNPs in 17 genes, significantly enhancing our grasp of the genetic predispositions to SAH. Particularly, mutations in TPO and PALD1 have been identified as novel risk factors, alongside an additional 25 genes that hint at key roles in extracellular matrix degradation and transcription factor signaling [31]. Similarly, WES has provided insights into cerebral small vessel disease (CSVD) and intracranial vertebral–basilar artery dissection (IVAD). For IVAD, an analysis of patients afflicted with isolated IVAD identified four known and seven novel variants in IVAD-related genes, as well as six variants in newly implicated genes [67]. In the context of CSVD, analysis of a Finnish patient cohort identified pathogenic variants in notable genes like NOTCH3, HTRA1, COL4A1, and COL4A2 in a significant proportion of patients, also unearthing variants associated with other neurological disorders [68]. These same gene variants have also been recognized as causative genes of CVDs worldwide.
In recent studies using WES to explore brain arteriovenous malformations (AVMs), significant genetic findings were uncovered. One study examined a Turkish family with three members having brain AVMs, and identified a ACVRL1 mutation in two siblings, suggesting that WES is exceptionally useful in cases of locus heterogeneity [69]. Additionally, research in AVM cases focused on identifying rare genetic mutations led to the discovery of 16 genes with unique mutations, with LRP2 and MUC5B being notable examples [70]. These findings highlight the potential of WES in understanding the genetics behind AVMs, offering new directions for research and treatment strategies in vascular diseases. The role of WES in CVDs has been illustrated in Fig. 2.
Fig. 2
Role of whole-exome sequencing in cerebrovascular diseases. ACOT4, Acyl-CoA Thioesterase 4; CHST14, Carbohydrate Sulfotransferase 14; CVDs, Cardiovascular Diseases; EDIL3, EGF-Like Repeats and Discoidin I-Like Domains 3; LRP2, Low-Density Lipoprotein Receptor-Related Protein 2; MUC5B, Mucin 5B, Oligomeric Mucus/Gel-Forming; NECD, Notch Endocrine Complex Delta (Typically referred to as NOTCH1 or Notch Receptor 1); NFX1, Nuclear Transcription Factor, X-Box Binding 1; NICD, Notch Intracellular Domain (part of the Notch signaling pathway); NNTM, Nicotinamide Nucleotide Transhydrogenase (Typically referred to as NNT); PALD1, Phosphatase Domain Containing, Paladin 1; PDE4DIP, Phosphodiesterase 4D Interacting Protein; PLOD3, Procollagen-Lysine,2-Oxoglutarate 5-Dioxygenase 3; SNPs, Single-Nucleotide Polymorphisms; TMEM132B, Transmembrane Protein 132B; TPO, Thyroid Peroxidase; TRPV3, Transient Receptor Potential Cation Channel Subfamily V Member 3
Neuro-oncological diseasesIn the realm of neuro-oncology, WES emerges as a transformative tool with significant implications for understanding and managing brain and spinal tumors. WES enables a comprehensive exploration of tumor genomics, addressing critical aspects such as tumor heterogeneity, the identification of diagnostic and therapeutic biomarkers, and the development of prognostic models. WES can be applied to two types of DNA: somatic and germline, each providing different insights into cancer genetics.
Somatic DNA refers to the genetic material found in the cells of the body, excluding the sperm and egg cells. Mutations in somatic DNA occur in specific cells during an individual's lifetime and are not inherited. These mutations can accumulate due to environmental factors, such as radiation and chemical compounds [71]. WES analysis of somatic DNA in tumors is essential for identifying mutations specific to cancer cells. This information helps researchers and clinicians understand the cancer's behavior and develop targeted therapies tailored to the genetic profile of the tumor [10].
In contrast, germline DNA is found in the sperm and egg cells and is inherited from one’s parents. Mutations in germline DNA are present in every cell of the body and can be transmitted to future generations. Germline mutations can predispose individuals to certain cancers, especially if the mutations include genes related to cancer development [72]. WES analysis of germline DNA is valuable for identifying inherited mutations that increase the risk of cancer. This information is essential for both assessing an individual's genetic predisposition to cancer, as well as for informing family members about potential risks [73].
Tumor heterogeneity and biomarkersWES offers unparalleled insights into the genomic landscape of brain and spinal tumors, unraveling the intricacies of tumor heterogeneity. Glioblastoma multiforme (GBM) is the most frequent and lethal primary brain tumor [74]. WES aided in the genetic and molecular profiling of patient-derived xenograft (PDX), leading to the discovery that PDXs recapitulate many key phenotypic and molecular features of patient tumors. The PDX models capture most molecular drivers, including TERT, EGFR, PTEN, TP53, BRAF, and IDH1, and preserve most genetic driver alterations, including EGFR amplification, found in patient tumors [74]. In a rare variation of GBM named giant cell GBM (gcGBM), WES revealed recurrent mutations of ATRX, PIK3R1, RB1 and SETD2 [75]. The application of WES in diffuse glioma (DG) also revealed TP53 and ATRX mutations, loss of function in PTEN and EGFR amplification, alongside CDKN2A/B deletion [24]. In pediatric patients with central nervous system (CNS) neoplasms, such as ependymoma, medulloblastoma and infiltrating astrocytoma, WES detected clinically pertinent Tier 1 (BRAF V600E, NTRK alterations, and C19MC amplification) and Tier 2 variants (BRAF fusion transcripts) [76]. The term “tier” refers to the classification of genetic variants based on their clinical relevance and potential impact on diagnosis, prognosis, and treatment. Tier 1 variants are those with strong clinical relevance, meaning they have significant implications for patient prognosis [76]. Tier 2 variants are defined as having moderate clinical relevance; they may have some evidence suggesting a potential impact on clinical outcomes, but this evidence is not conclusive. Tier 3 variants consist of variants of unknown significance that do not meet the criteria for Tiers 1 or 2 [76].
The application of WES in the study of brain metastases (BM) has yielded significant insights. Comparative analyses between BMs and primary tumors have uncovered noteworthy observations. Specifically, BMs exhibit a higher tumor mutational burden, characterized by elevated mutational signatures associated with homologous recombinant deficiency (HRD) and mismatch repair deficiency (MMRD) [77, 78]. In the context of colorectal cancer, frequent BM-specific mutations have been identified, encompassing DDR, SCN7A, SCN5A, SCN2A, IKZF1, and PDZRN4 [77]. Conversely, in BM originating from triple-negative breast cancer (TNBC), TP53 mutations are prevalent, constituting the most frequently mutated gene in this context [78]. Moreover, lung cancer BM have exhibited mutations in KMT2C and AHNAK2 [79]. These findings offer the potential for distinguishing the origin of BM, be it from breast cancer, lung cancer, or colorectal cancer. Furthermore, the novel metastasis-related mutations identified through WES hold promise as biomarkers for diagnostic and targeted therapeutic interventions.
WES has contributed significantly to advancing our comprehension of the genetic profile of sporadic vestibular schwannoma (VS), a benign tumor characterized by associated morbidities and diminished quality of life [32]. The findings underscore the marked heterogeneity within the genetic landscape of VS. Despite this diversity, a predominant pattern emerges wherein the majority of samples exhibit mutations either in the NF2 gene or in genes closely associated with NF2 [32]. Notably, the study establishes that Gamma Knife radiosurgery (GKRS) does not correlate with an elevated incidence of mutations in the context of VS [32]. Unraveling the exomic landscape allows for a precise characterization of molecular signatures, guiding the identification of specific pathways implicated in tumorigenesis.
These results collectively enhance our understanding of the genetic intricacies of brain tumors and provide valuable insights into the impact of interventions such as GKRS on its mutational landscape. By leveraging insights into tumor heterogeneity and biomarkers from WES, clinicians can tailor treatment strategies with heightened precision, facilitating the implementation of personalized therapeutic regimens. The role of WES in neuro-oncology has been illustrated in Fig. 3.
Fig. 3
Role of whole-exome sequencing in neuro-oncological conditions. AHNAK2, AHNAK Nucleoprotein 2; ATRX, Alpha Thalassemia/Mental Retardation Syndrome X-Linked; BBB, Blood–Brain Barrier; BRAF, B-Raf Proto-Oncogene, Serine/Threonine Kinase; C19MC, Chromosome 19 MicroRNA Cluster; CNS, Central Nervous System; EGFR, Epidermal Growth Factor Receptor; gcGBM, Giant Cell Glioblastoma; GKRS, Gamma Knife Radiosurgery; IDH1, Isocitrate Dehydrogenase 1; KMT2C, Lysine Methyltransferase 2C; NTRK, Neurotrophic Receptor Tyrosine Kinase; PDX, Patient-Derived Xenograft; PIK3R1, Phosphoinositide-3-Kinase Regulatory Subunit 1; PTEN, Phosphatase and Tensin Homolog; RB1, Retinoblastoma 1; SCN2A, Sodium Voltage-Gated Channel Alpha Subunit 2; SCN5A, Sodium Voltage-Gated Channel Alpha Subunit 5; SCN7A, Sodium Voltage-Gated Channel Alpha Subunit 7; SETD2, SET Domain Containing 2; TERT, Telomerase Reverse Transcriptase; TP53, Tumor Protein p53; V600E, Valine replaced by Glutamic acid at position 600; WES, Whole-Exome Sequencing
留言 (0)