Reagents and antibodies
Reagents in this study were obtained as follows. Antimycin A (#1397-94-0), BODIPY 493/503 (#D2191), Mitochondrial Superoxide Indicators (MitoSOX, #M36008), and Hoechst (#62249) from Thermo Fisher Scientific (Boston, USA); Oliec acid (#O1383), Palmitic acid (#P9767), and RIPA buffer (#R0278) from Sigma-Aldrich (Missouri, USA); Albumin BovineV (fatty acid-free; #A8850) from Solarbio (Beijing, China); Dihydroethidium (DHE; #S0063), ROS Assay Kit (#S0033S), Lipid Peroxidation MDA Assay Kit (#S0131), Catalase Assay Kit (#S0051), Puromycin (#ST551), and DAPI (#C1005) from Beyotime (Shanghai, China); Oligomycin (#S1479), ADP (#S9368), Succinic acid (#S3791), Glutamic acid (#S6266), Malic acid (#S9001), FCCP (#S8276), Rotenone (#S2348), and Dorsomorphin (#BML-275) from Selleck Chemicals (Houston, TX, USA); Rhod-2 AM (#HY-D0989) from MedChemExpress (New Jersey, USA). Penicillin, Streptomycin, and trypsin-ethylenediaminetetraacetic acid from Genom Biotechnology (Shanghai, China); and ATP Luminescent kit (#40210ES10) from Yeasen Biotechnology (Shanghai, China). All antibodies in this study are listed in Table S1.
Animals, diets, and experimental design
Six-week-old Male C57BL/6J wild-type (WT) mice (Shanghai Slack laboratory Animal Co., Ltd., Shanghai, China) lived in an SPF condition. Mice had unrestricted access to food and liquids with a 12-hour light-dark cycle. Mice were randomly divided into 4 groups (n = 9–15 mice/group). Mice were given either a normal chow diet (NCD) or a choline-deficient, L-amino acid-defined, high-fat diet (CDAHFD; Research Diets, USA) for 8 weeks [29, 30]. Conditional MCU knockdown mice were using adeno-associated virus serotype 8 (AAV8). For the CRISPR/Cas9 studies, All-in-One AAV8 encoding small-guide RNA (sgRNA) targeting MCU or scrambled sgRNA were provided by Hanyin Biotechnology Limited Company (Shanghai, China) (Additional file 1: Fig. S1). C57BL/6J mice were injected with either a mixture of AAV8-sgMCU or AAV8-sgCtrl at the beginning of CDAHFD-feeding. After 8 weeks, except for 3 mice in each group for use to isolate primary hepatocytes, all mice were sacrificed. Liver samples were stored at -80 °C or fixed in freshly prepared 4% paraformaldehyde.
To further verify the expression of MCU in the liver, a different diet-induced MASH mouse model was induced by feeding a high-fat/calorie diet (HFCD) for 16 weeks along with high carbohydrates (HC: 55% fructose/45% glucose) in drinking water [31]; and mice with NCD serves as the control (n = 8 mice/group). The Animal Care Committee of Zhongshan Hospital, Fudan University, accepted the guidelines for the care and use of laboratory animals, which were followed in all animal research (No. 2019 − 186).
Cell cultures and treatments
From the specified animals, primary hepatocytes were extracted using the collagenase perfusion procedure that has been previously published [6]. In summary, newly extracted hepatocytes were cultivated in RPMI medium 1640 supplemented with 1.0 g/L glucose, 10% fetal bovine serum (FBS), 100 units/mL penicillin, and 100 mg/mL streptomycin at 37 °C in a humidified chamber. The hepatocytes were seeded into collagen I-coated petri plates. Six hours after plating, the medium was changed, and the cells received the appropriate treatment [32]. In this study, AML12 or primary hepatocytes were induced by oleic acid and palmitic acid (OPA) as the model of saturated fatty acid-induced lipotoxicity [33]. Briefly, 200 µM OA and 100 µM PA were first conjugated to bovine serum albumin (BSA) and then added to the medium to induce oxidative stress and hepatocellular lipotoxicity.
AML12 hepatocytes were transfected with different short hairpin (sh) RNAs as follows: non-targeted scrambled control shRNA (shScr) and 2 groups of shMCU (#1 Sequence 5’~3’ CACGTTTCGACCTAGAGAA and #2 Sequence 5’~3’ GGAGAAGGTACGAATTGAA). The shMCU and shScr lentiviral transduction particles were provided by Hanyin Biotechnology (Shanghai, China). The shMCU and shScr were synthesized and the inserted into a U6-MCS-CMV-Puro vector, and the empty vector was packaged as negative control. Then the stable transfectants were screened and cultured in medium containing puromycin. MCU knockdown was confirmed by qRT-PCR and western blotting respectively (Additional file 1: Fig. S2A and B). We finally selected shMCU (#1) with the highest efficiency for the construct of the shRNA.
Primary hepatocytes were transfected with siRNA for MCU to transiently knock down MCU. Non-targeted scrambled control siRNA (siScr) and 3 groups of siMCU were transfected in AML12 cells to verify the knockdown efficiency of siMCU and select the most efficient one. The siMCU and siScr were provided by Genepharma (Shanghai, China). The produced oligo sequences are displayed in Table S2. MCU knockdown efficiency was verified by qRT-PCR and western blotting, respectively (Additional file 1: Fig. S2C and D). The highest knockdown efficiency siMCU (#1) was finally selected to transfect primary hepatocytes.
The cDNA encoding mouse MCU (1053 bp) was produced by qRT-PCR and was subcloned into the EcoRI site of the pEX-1 (pGCMV/MCS/EGFP/Neo) expression vectors (GenePharma, Shanghai, China). AML12 hepatocytes were transfected with mouse MCU cDNA plasmids (5 µg) or the empty vector pEX-1 as a negative control for 48 h using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s protocols. The effectiveness of genetic overexpression was confirmed by qRT-PCR and western blotting (Additional file 1: Fig. S2E and F), respectively. Then the transfected cells were incubated in DMEM/F12 medium with OPA or vehicle treatment for 24 h, cells were collected and analyzed for further testing.
Whole-transcriptome amplification and RNA-sequencing analysis
Employing Trizol Reagent (Thermo Fisher), total RNA was extracted from liver tissues and utilized for RNA-seq analysis. On an Apollo Library Prep System (Illumina, San Diego, CA), library preparation was carried out using the TruSeq Stranded mRNA Sample Prep Kit (Takara, Shiga, Japan). Liver RNA-seq was performed on an Illumina Novaseq™ 6000 platform in 2 × 150 bp paired-end sequencing (LC-Bio Technology, Hangzhou, China). We used HISAT2 (https://daehwankimlab.github.io/hisat2/, version: hisat2-2.2.1) package to map the clean reads to each gene and normalized the raw data to Fragments Per Kilobase of exon model per Million mapped fragments for subsequent analyses. Bioinformatics analyses were performed as previously described [34]. Differentially expressed genes (DEGs) were identified with the limma package, which implements an empirical Bayesian approach to estimate gene expression changes by using the moderated t-test [35]. |log FC| > 0.5 and P < 0.05 were considered cutoff criteria to screen for DEGs. Functional enrichment analyses of the detected DEGs were performed with the clusterProfiler package. GO and Kyoto Encyclopedia of Genes and Genomes (KEGG) terms were identified with a cutoff of P < 0.05. We also identified pathways that were upregulated or downregulated in NASH livers from 2 groups [36]. Gene sets for analysis were obtained from the MSigDB database; and enrichment analyses of DEGs were performed using Metascape. After finding the pathway we use the Cytoscape (Ver3.9.1) to search for differential genes. Finally, the mechanism diagram was rendered using Biorender software.
Human liver specimens
Human liver specimens were taken from MASLD/MASH patients who had undergone surgical liver resection or liver biopsy; and healthy liver tissues were taken from the unaffected portion of the liver of patients having surgery to repair a hepatic hemangioma or trauma. All liver samples are obtained from Shanghai Public Health Clinical Center that approved by the ethics committees of Shanghai Public Health Clinical Center (2022-S104-01). Written informed permission was provided to all participants in accordance with the Helsinki Declaration. Table S3 displays data from laboratory and clinical studies.
Histopathology and immunohistochemistry
For histopathological and immunohistochemical (IHC) assessment, paraffin-embedded formalin-fixed liver sections were stained with H&E, Masson’s trichrome, or sirius red according to standard protocols. Liver histology was blinded reviewed by two pathologists to conduct steatosis grading and NAFLD activity score (NAS) as previously described [32]. Additionally, liver fibrosis and fat content were also assessed blindly using ImageJ software based on Sirius red and H&E-stained sections, respectively.
IHC staining was done according to our earlier instructions [32]. In summary, sections with a thickness of 5 μm were dewaxed, hydrated, and then heated to induce antigen retrieval. After that, slides were blocked and incubated with primary antibodies against α-SMA, MCU, Cyto c, F4/80, anti-4-HNE, and anti-cleaved Caspase-3 over night at 4 °C. The primary and secondary antibodies used are listed in Supplemental Table S1. Sections were subsequently incubated with HRP-conjugated secondary antibodies and finally examined under a light microscope to detect. Immunostaining signaling was quantified at a predetermined threshold using free NIH ImageJ 1.49 for 6-9 slides/group, 5 fields per slide.
Oil red O and BODIPY staining
Lipid accumulation was analyzed using an Oil red O (ORO) staining kit (ScienCell, California, USA, #0843) as described in the manufacturer’s protocol. AML12 or primary hepatocytes were fixed with 4% paraformaldehyde for 10 min. Then, cells were stained with freshly diluted 0.2% ORO solution in a 37 °C incubator for 10 min. Plates were counterstained with hematoxylin to stain cell nuclei. As for liver sections, ORO staining in sections from frozen liver tissue was conducted as described previously [30, 32]. The positive area was assessed from randomly selected 6 fields of ×200 magnification per slide and quantified with NIH ImageJ 1.49 software.
For BODIPY staining, the cells were treated and fixed the same with ORO staining. Then the fixed cells were stained with BODIPY 493/503 dye under RT for 10 min and protected from light, and the nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI). The stained lipid droplets were observed under a confocal microscope (FV3000, Olympus). The areas of the lipid droplets in different groups were analyzed using NIH Image J 1.49 software.
Western blot
Western blot analysis was performed according to standard protocols as previously described [30, 32]. Cells and liver tissues homogenized in RIPA buffer were centrifugated at 4 °C, and protein concentrations were assessed by micro-BCA assay kit (Beyotime, Shanghai, China). Protein (10–20 µg) was dissolved by electrophoresis on 12.5% SDS-PAGE gels and transferred to polyvinylidene fluoride membranes. The membranes were blocked and incubated overnight at 4 °C with primary antibodies against MCU, β-Actin, Caspase-3, cleaved Caspase-3, α-SMA, CPT1A, AMPK, p-AMPK, YAP1, p-YAP1, LATS1/2, p-LATS1/2, MOB1, and p-MOB1. Followed by washing 3 times, the membranes were incubated with peroxidase-conjugated secondary antibodies under RT. The primary and secondary antibodies used are listed in Table S1. COX IV or β-actin was used as a loading control. Bands were visualized with ECL™ Western Blotting Detection Reagents (EpiZyme, Shanghai, China), and the optical density of the bands were determined using the NIH ImageJ software.
RNA isolation and quantitative real-time PCR
Total RNA was extracted from cells or liver tissue using Trizol reagents (Takara) according to the manufacturer’s instructions. For qRT-PCR, cDNAs were reverse transcribed using GoScript™ Reverse Transcription Mix and Random Primers (Promega). Relative quantitative gene expression levels were measured by qRT-PCR using Promega SYBR Green mix kit on QuantStudio 5 System (Applied Biosystems, CA). β-actin was used as an internal control, and the relative expression of target genes was calculated using the 2−ΔΔCT method. All primers used are listed in Table S4.
TUNEL staining
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining was utilized to identify apoptotic cells in liver sections and in vitro AML12 hepatocytes. Formalin-fixed and paraffin-embedded liver sections were stained using the DeadEnd™ Fluorometric TUNEL System (#G3250; Promega, Wisconsin, USA) following the manufacturer’s instructions. Olympus BX43 microscope was applied to view the slides after they had been counterstained with DAPI. Then recordings were made using an Olympus FV3000 confocal microscope.
Flow cytometry analysis
Apoptosis/necrosis was performed using a FITC Annexin V Apoptosis Detection Kit I (#556547; Becton Dickinson Biosciences, USA) according to the manufacturer’s protocol. Briefly, AML12 cells were treated as indicated and harvested, collected, and resuspended in 100 µL of binding buffer and stained with annexin V FITC (5 µL) and propidium iodide (PI) staining solution (5 µL) for 20 min at RT in the dark. Samples were chilled before testing, and flow cytometry was employed to detect cellular apoptosis. Data were analyzed using the Flow Jo software (V10.8.1).
Determination Caspase3/7 activity
Caspase-3/7 activities in cells were measured using the Caspase-3/7 Activity Apoptosis Assay Kit *Green Fluorescence (Sangon Biotech, Shanghai, China) according to the manufacturer’s protocol as described previously [32]. Briefly, cultured with or without OPA for 24 h, AML12 cells were lysed with buffer containing caspase substrate TF2-DEVD-FMK and incubated for 1 h at RT. Caspase3/7 activities were measured using FlexStation3 multifunctional microplate reader (Molecular Devices, California, USA) with excitation wavelength at 490 nm and emission wavelength at 525 nm.
Assessment of hepatic oxidative stress and lipid peroxidation
As previously mentioned, the Lipid Peroxidation MDA Assay Kit was utilized in accordance with the manufacturer’s directions to measure the amount of lipid peroxidation in the mice’s livers. The manufacturer’s procedure was followed to evaluate the hepatic Catalase activity using the Catalase Assay Kit. The results were expressed as micromoles of decomposed H2O2 per minute per milligram of liver protein. By measuring the amounts of superoxide anion using DHE staining, as previously described, oxidative stress was identified in frozen and unfixed liver tissue slices. Summing up, cryosections of the livers from each mouse group were incubated for 30 min at 37 °C with 25 µM DHE in a light-protected, humidified chamber. After that, they were rinsed three times with PBS. DHE fluorescence was captured using an Olympus BX43 fluorescence microscope.
Hydroxyproline content
Hydroxyproline content of the livers was measured using the hydroxyproline detection kit (#A030-2-1; Jiancheng, Nanjing, China) according to the manufacturer’s instructions. Briefly, liver tissue (40 µg) was hydrolyzed with 1 ml of hydrolysate, and bathed in water at 95 °C for 20 min. After cooling the tubes to RT in running water, we added 10 µL of pH indicator, solution A, and solution B to each tube sequentially to adjust the pH value to 6.0-6.8. The liquid in the tubes was then diluted to a volume of 10 mL. 4 mL of diluted hydrolysate was mixed with the required amount of activated carbon, and the mixture was then centrifuged at 3,500 rpm for 10 min to collect the supernatant. Sample and standard were used to measure the amount of hydroxyproline in 100 µL of supernatants at a wavelength of 550 nm. Using the formula, collagen levels were estimated from the product of hydroxyproline content (µg/g liver).
Measurement of ALT and AST in serum
Blood was collected and stored at -80 °C until use. Serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were evaluated with a commercial assay kit according to the manufacturer’s recommendations (Nanjing Jiancheng Biological Technology, Nanjing, China). Liver enzyme activities were shown in international unit per liter (U/L).
Measurement of intracellular ROS and mitochondrial superoxide levels
Cellular ROS generation was measured using the ROS Assay Kit as previously described. Briefly, after being stained with 10µM DCFH-DA for 20 min at 37 °C, the incubated cells were washed and lysed in PBST. Finally, fluorescence was measured at 495 nm/530 nm (Ex/Em) under Olympus BX43 microscope or Flow cytometry (FACS Arial, Becton Dickinson). Staining for mitochondrial superoxide (miROS) was performed using the MitoSOX Red indicator following the manufacturer’s instructions as previously described.
Transmission electron microscopy
For electron microscopy, Kreb’s solution and a 0.15 M cacodylate buffer containing 2.5% glutaraldehyde, 2% paraformaldehyde, and 2 mM CaCl2 were both injected into mice, followed by 5 min of heating to 37 °C. After perfusion, liver tissue was carefully removed and once again fixed for the night at 4 °C. The tissues were washed 3 times in 0.15 M cacodylate buffer, then post-fixed with 1% osmium tetroxide and 0.3% potassium ferrocyanide in 0.15 M cacodylate buffer with 2 mM CaCl2. Following washing 3 times in ddH2O, liver tissue was en bloc stained with 2% aqueous uranyl acetate overnight at 4 °C and subsequently dehydrated in a Spurr’s resin (30%, 50%, 70%, and 100%) mixed with 100% ethanol, embedded in fresh 100% Spurr’s resin in silicon molds, and polymerized at 60 °C for 48 h. Following polymerization, the resin blocks were faced, 70 nm ultrathin sections were cut on a Leica EM UC7 ultramicrotome (Leica-Microsystems, Vienna, Austria), picked up on copper formvar/carbon support film grids (PN FCF100H-Cu, Electron Microscopy Sciences, Hatfield, PA), post-stained with 2% uranyl acetate for 15 min, followed by Reynolds lead citrate and imaged on 5–6 photos per sample at a magnification of 2000 were taken.
ATP measurement
Hepatic ATP content was measured using the ATP Luminescence Kit (#40210ES10; Yeasen Biotechnology, Shanghai, China) according to the manufacturer’s instructions [32]. Briefly, after homogenization in RIPA lysis buffer, 20 mg of liver tissue was used. For intracellular assessment, 5 × 105 cells/well were washed and then lysed in 100 µL buffer after treatment with OPA or vehicle. The lysate was finally centrifuged at 12,000×g for 3 min to collect the cell supernatant. Samples were added to a 96-well plate and then incubated with 100 µL of the test working fluid at RT in the dark for 2 s. Absorbance was read the Luminescene using FlexStation3 multifunctional microplate reader (Molecular Devices). ATP content was calculated based on a standard curve generated at the same time.
Measurement of mitochondrial membrane potential (MMP)
The MMP was determined using the JC-1 Mitochondrial Membrane Potential Assay Kit (#10009172; Cayman, Michigan, USA) according to the manufacturer’s instructions [32]. Briefly, AML12 cells were seeded in a 12-well Glass Bottom Plate (Cellvis) and treated respectively. After being washed with PBS, cells were incubated with JC-1 for 30 min at 37 °C. Nuclei were counterstained with Hoechst. In a nutshell, Alexa Fluor 594 channel and Alexa Fluor 488 channel were adopted, respectively, for the detection of JC-1 in its J-aggregate form (red fluorescence) and monomeric form (green fluorescence). The ratio between red and green fluorescence was calculated to indicate the MMP.
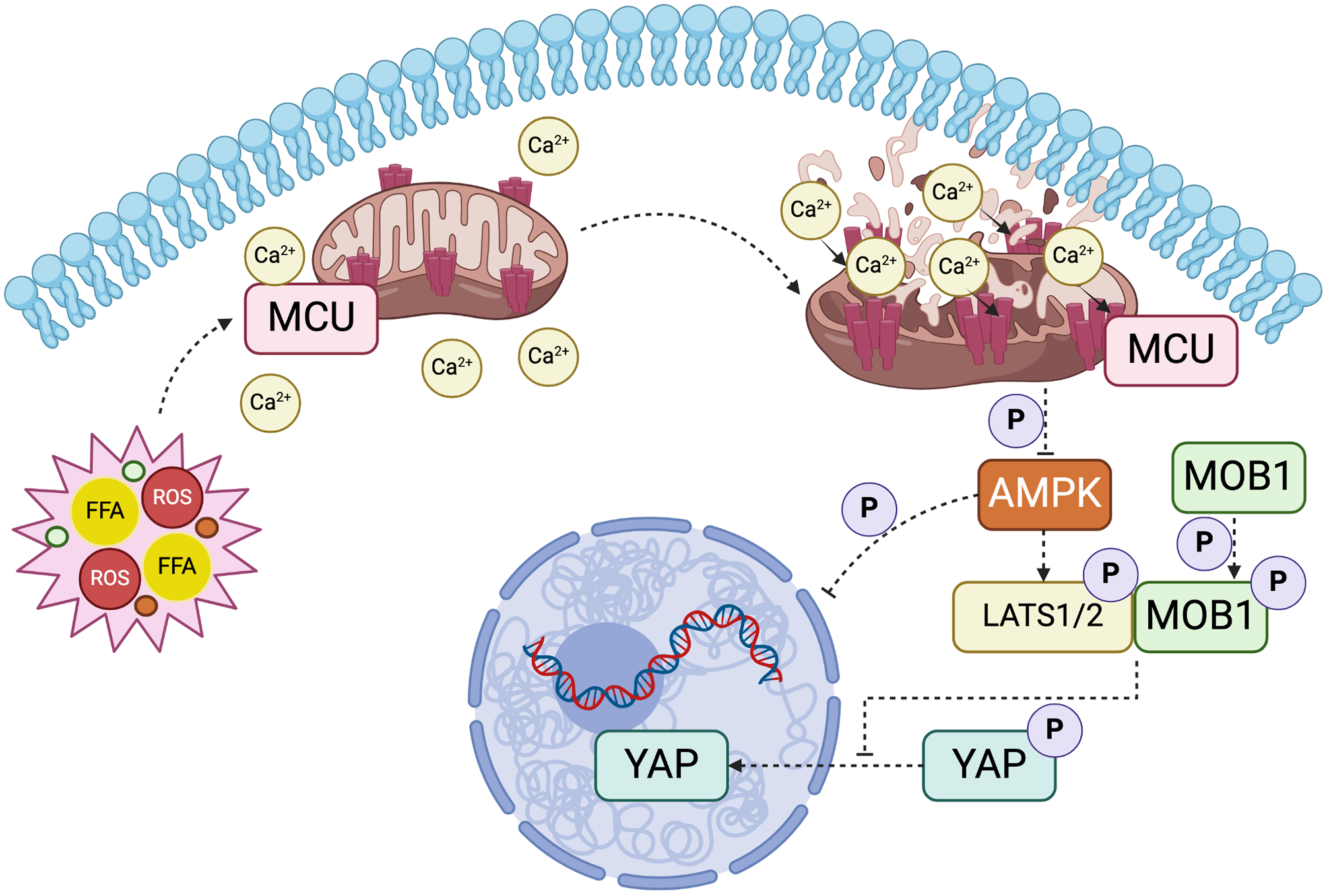
Measurement of mitochondrial Ca2+ concentration
Mitochondrial Ca2+ concentration was assessed as described previously [24]. Briefly, AML12 hepatocytes cultured in a 12-well Glass Bottom Plate (Cellvis) or confocal dishes (Biosharp Life Sciences) were treated separately. After being washed with PBS, cells were loaded with Rhod-2 AM (5 µM) for 30 min at 37 °C. The fluorescence of cells was observed under Opera Phoenix High-Content Screening System (PerkinElmer, CT) with Alexa Fluor 488 channel or a confocal microscope (Olympus FV3000) with Alexa Fluor 594 channel. Data were analyzed using ImageJ software.
Measurement of mitochondrial oxygen consumption rate
Following the manufacturer’s instructions, liver mitochondria were separated from the appropriate animals (five mice per group) at the end of eight weeks using a tissue mitochondria isolation kit (Beyotime Institute Biotechnology) [32]. In a nutshell, 50 mg of liver samples were diced and homogenized (1:10 w/v) in cold mitogen buffer. After that, portions of the plasma membrane and nuclei were spun down by centrifuging the tissue homogenate. To get rid of impurities, the supernatant was filtered and centrifuged. In order to extract the mitochondria, we also extracted the cytoplasmic proteins from the supernatant. Next, the pellet was immersed in 100 µl of mitochondrial buffer.
Mitochondrial function was measured by oxygen consumption rate (OCR) using Seahorse XF96 Analyzer (Agilent Technologies, California, USA) as previously described [37]. Briefly, hepatic mitochondria were incubated in a mitochondrial assay solution. Substrate stocks containing 0.5 M succinic acid, 0.5 M malic acid, 0.5 M glutamic acid, 0.5 M pyruvic acid, and 1.0 M ADP, and adjusted to pH 7.2. Respiration reagent stocks were used: FCCP (10 mM), oligomycin (5 mg/mL), rotenone (2 mM), and antimycin A (40 mM). 2 µg mouse liver mitochondria work well in the Agilent Seahorse XF96 begin in a coupled state with substrate present. ADP, oligomycin, FCCP, Rotenone, and Antimycin were sequential additions to the reaction system. State 3 initiated with ADP, State 4 induced with the addition of oligomycin (State 4o), and FCCP-induced maximal uncoupler-stimulated respiration (State 3u) were sequentially measured, allowing respiratory control ratios. Respiratory control ratio (RCR) was defined as the ratio of state 3 respiration to state 4o. Data were collected using Agilent Seahorse Wave 2.6.1 Desktop software and analyzed by GraphPad Prism version 9.
Statistical analysis
All data are presented as means ± standard deviations (SD), and the data have passed the normality and equal distribution test. Statistically significant differences between two independent groups were made by unpaired two-tailed Student’s t-test. Comparisons among multiple groups were performed by one-way ANOVA with Tukey’s post hoc honest significant difference test or two-way ANOVA followed by Tukey’s or Sidak’s multiple comparisons test. Statistical analyses were performed using GraphPad Prism 9.0 (San Diego, CA, USA). For all comparisons, significance is indicated as *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001; ns indicates not significant.



留言 (0)