Materials
Cetyltrimethylammonium chloride (CTAC), bis[3-(triethoxysilyl) propyl] tetrasulphide (BTES), triethanolamine (TEA), tetraethyl orthosilicate (TEOS), ethanol, ammonia aqueous solution (NH3·H2O, 25 wt%), and collagenase were sourced from Sigma-Aldrich (MO, USA). (3-mercaptopropyl) trimethoxysilane (MPTES), sodium sulfide nonahydrate (Na2S·9H2O), 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), sodium citrate, L-glutathione (GSH), 2-iminothiolane (Traut’s reagent), copper chloride dihydrate (CuCl2·2H2O), and disulfiram (DSF) were obtained from Aladdin Co., Ltd. (Shanghai, China). Additionally, the Membrane and Cytosol Protein Extraction Kit, blotting grade, primary antibody dilution buffer, DCFH-DA, fluorescein isothiocyanate (FITC) and 4’6-diamidino-2-phenylindole (DAPI) were acquired from Beyotime (China).
Cell lines and animals
The BxPC-3 cell line, which originates from human pancreatic cancer, was procured from Procell (Wuhan, China). These cells were maintained in an RPMI 1640 medium containing 10% FBS at 37 °C within a humidified incubator with 5% CO2. Male BALB/c nude mice, aged 6–8 weeks, were sourced from the Shanghai Silaike Laboratory Animal Limited Liability Company. All animal experiments were conducted in accordance with the guidelines set by the National Institutes of Health (NIH, USA) and were approved by the Animal Experiment Committee of Zhejiang University (Approval Year: 2024, No. 040). To generate the BxPC3 tumor-bearing mouse model, 2.0 × 106 cells were subcutaneously injected into the left thigh of male BALB/c nude mice. The volume of the tumor (V) was determined using the formula V = a²×b/2, where ‘a’ represents the shortest length (mm) and ‘b’ represents the longest length (mm) of the tumor.
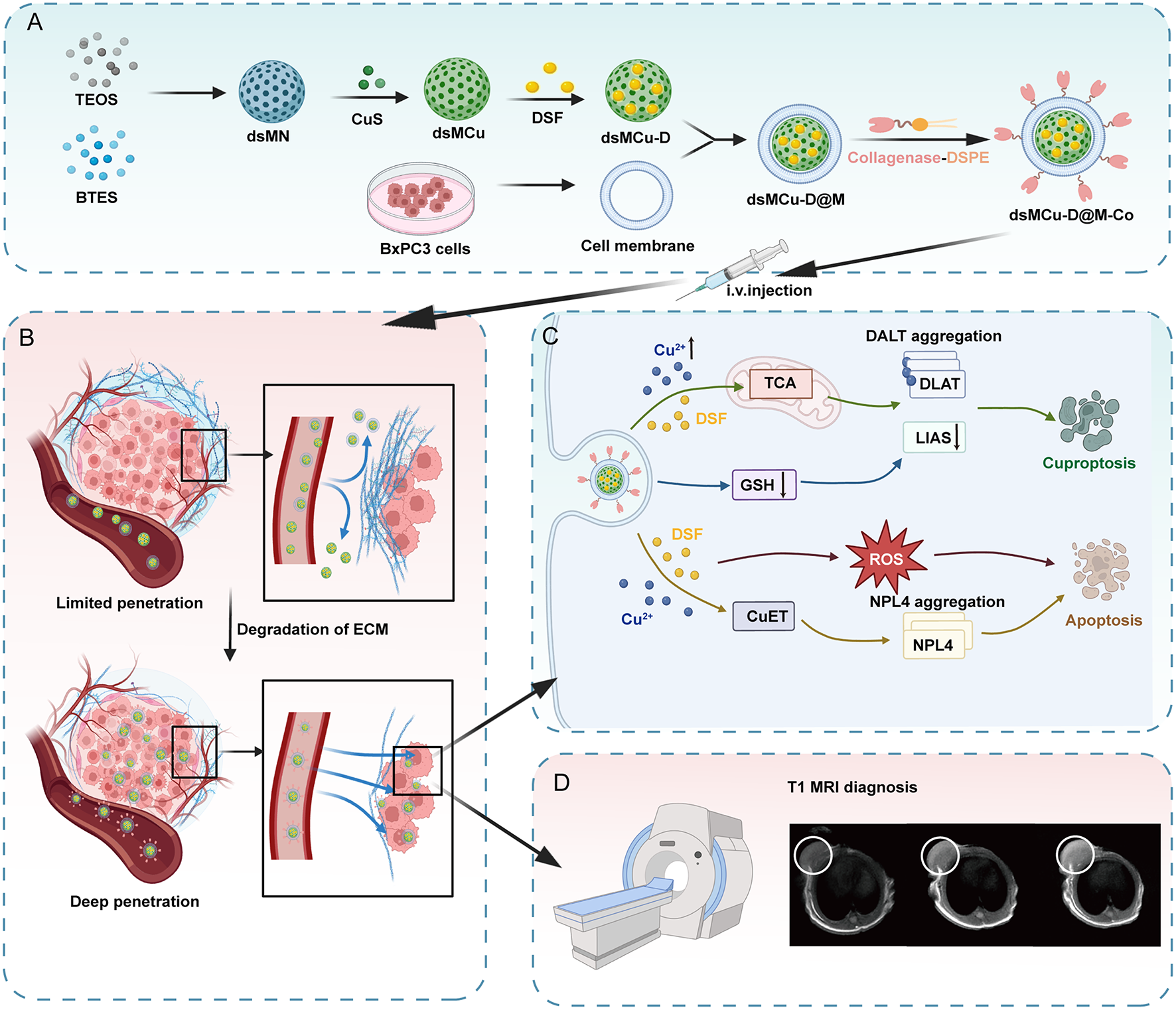
Synthesis of dsMNs
Initially, TEA (6 g, 10 wt%) and CTAC (40 g, 10 wt%) aqueous solutions were mixed and stirred at 85 °C for 15 min. TEOS (2 ml) was then added dropwise and allowed to react for 1 h. Following this, a mixture of TEOS (1 ml) and BTES (2 ml) was added, and the reaction was continued for an additional 4 h. The resulting products were washed thrice with ethanol. Then the resulting products were dispersed in 50 mL methanol containing 1 wt% NaCl and heated at 65 °C, to remove the template CTAC. This step was repeated at least three times, each time at least 12 h to ensure the template was removed completely. The final dsMN products were obtained using an ammonia-assisted selective etching strategy, which involved reaction of the NPs with an aqueous solution of ammonia (25 wt%) for 2.5 h at 90 °C. After the solution cooled to room temperature, the final dsMN products were acquired by centrifuging and subsequently washing three times with water [31].
Synthesis of dsMCu
dsMCu were synthesized according to the classical synthesis method. First, 4 mL of a 0.05 M Na2S·9H2O aqueous solution was added to 200 mL of a mixed solution containing 1 mmol CuCl2·2H2O and 0.68 mmol sodium citrate. The mixture was stirred at ambient conditions for 30 min, followed by heating to 90 °C and maintaining this temperature for 10 min. This process yielded a dark-green copper sulfide (CuS) NP solution which should be transferred to an ice bath immediately. Next, dsMNs (15 mg) were dispersed in 40 mL ethanol, and a mixture of 0.15 mL MPTES and 0.2 mL NH3·H2O (25 wt%) was added to it. After stirring overnight, the resulting dsMN-SH was collected via centrifugation and washed thrice with ethanol. Next, 60 mL of the CuS solution and 30 mg dsMN-SH were re-dispersed in 100 mL water and stirred for 2 h in ice bath. The dsMCu NPs were obtained via centrifugation and then washed three times with water [32].
Loading DSF into dsMCu (dsMCu-D)
For incorporating DSF molecules into dsMCu NPs, 10 milligrams of dsMCu NPs were dispersed in 2 milliliter of ethanol solution containing 20 milligrams of DSF, followed by overnight vigorous stirring. Subsequently, the dsMCu-D NPs composite was obtained through centrifugation and washed thrice with ethanol for subsequent applications. Then, the encapsulated dsMCu-D NPs samples are placed into the thermogravimetric analyzer and heated within a specific temperature range. At different temperatures, both the drug and the carrier undergo mass changes. The encapsulation efficiency and drug loading values were calculated by observing the variations in the sample mass.
Extraction and preparation of BxPC-3 cell membrane-coated nanoparticles
BxPC-3 cell membranes were isolated using a membrane protein extraction kit from Beyotime (China), following the manufacturer’s guidelines. Initially, BxPC-3 cells were cultured and suspended in reagent A for membrane protein extraction, followed by incubation on ice for 20 min. The cells were then lysed by repeated freezing in liquid nitrogen and subsequent thawing at room temperature until complete lysis was achieved. After centrifugation at 800 g and 4 °C for 15 min to remove intact cells and nuclei, the lysate was further centrifuged at 15,000 g and 4 °C for 40 min to separate out the cell membranes. The supernatant was discarded, and the pellet containing the BxPC-3 cell membranes was collected. To prepare dsMCu-D@M nanoparticles, dsMCu-D was combined with BxPC-3 cell membranes using sonication and then extruded through a 200 nm polyethylene terephthalate membrane (LiposoFast, Avestin, Canada). The resulting dsMCu-D@M nanoparticles were harvested by centrifugation and washed with water [33].
Synthesis of dsMCu-D@M-Co
For the synthesis of dsMCu-D@M-Co, collagenase-modified DSPE-Col was synthesized first. Traut’s reagent in a 10 mg/mL concentration and collagenase in a concentration of 10 mg/mL were combined and stirred at room temperature for one hour to synthesize thiolated collagenase (SH-Col). The excess Traut’s reagent was removed by dialysis. Then, DSPE-PEG-MAL at 15 mg/mL was mixed with SH-Col at a concentration of 7.5 mg/mL to synthesize DSPE- PEG-Col. The dialysis removed any unbound DSPE- PEG-MAL. The addition of this DSPE- PEG-Col into the dsMCu-D@M and incubation with it at room temperature for one hour allowed the lipids to fuse with the cell membrane [34]. The unconjugated DSPE-Col was dialyzed out. Finally, dsMCu-D@M-Co was centrifuged to obtain the product, which was washed with water.
Collagenase activity study
The residual activity of collagenase was determined first on gelatin. A gelatin solution (30 mg/mL) was incubated at 37 °C with DSPE-Col, free collagenase, dsMCu-D@M, and dsMCu-D@M-Co. After 24 h of incubation, each sample was cooled at four °C for 30 min before being documented with a digital camera. At the same time, activity was assessed by determining collagenase activity using the Collagenase Activity Colorimetric Assay Kit (Sigma-Aldrich, St. Louis, MO, USA). Collagen was mixed with PBS that included FITC (10 µg/mL) and put in ice. Next, the mixture was added to a 24-well plate and allowed to solidify at room temperature for 20 min. When gel appeared, it was softly washed till the fluorescence of the supernatant became zero [35]. After that, 200 µL of PBS, dsMCu-D@M, dsMCu-D@M-Co, and collagenase were applied on the surface of the gel. After 24 h of incubation at 37 °C, the fluorescence in the supernatant was measured.
Characterization
The particle size and zeta potential were determined using a Litesizer500 particle analyzer (Anton-Paar, Austria). The structural features of the dsMN, dsMCu, dsMCu-D@M, and dsMCu-D@M-Co nanoparticles were investigated using the transmission electron microscopy (TEM) JEM-1400fash JEOL, Japan. The surface area and pore size distribution were determined by the Brunauer-Emmett-Teller (BET) and Barrett-Joyner-Halenda (BJH) techniques. Ultraviolet absorption spectra were determined on a UV-2600 spectrophotometer by SHIMADZU, Japan. Fourier transform infrared (FTIR) spectra were recorded using a 330FT-IR spectrometer produced by Thermo Fisher Scientific, USA. The copper content of the samples was determined by inductively coupled plasma mass spectrometry with a Nexion 300X instrument from PerkinElmer, USA. The loading percentage of the drug was determined using a thermogravimetric analyzer, and the drug loading was calculated by dividing the weight of DSF by the total weight of the drug-loaded product and multiplying the whole by 100%.
Collagen degradation in vitro
BxPC-3 cells were seeded in an 8-chamber slide overnight for adhesion. The cells were washed with PBS, and a fresh medium containing different NPs was added. After two PBS rinses, the cells were fixed with 4% formaldehyde for 10 min. Collagen was immunostained using an Anti-Collagen I antibody according to the manufacturer’s instructions, and imaging was performed using confocal laser scanning microscopy (CLSM).
Assessing cellular uptake
BxPC-3 cells were plated onto 8-chamber slides (Cellvis, USA) and incubated overnight at 37 °C with 5% CO2. Upon reaching about 70% confluence, cells had their old medium substituted with fresh media, which included FITC-labeled dsMCu-D, dsMCu-D@M, and dsMCu-D@M-Co. After 2–8 h, the cells were washed three times with PBS. After fixing the cells with a 4% paraformaldehyde solution for 30 min, actin-tracker and DAPI were used to label F-actin and the nucleus, respectively. Finally, images were captured using a Nikon microscope (Nikon Eclipse Ti-S, USA). Cells undergoing various treatments were also examined using bio-TEM.
Cytotoxicity analysis in vitro
BxPC-3 cells were allocated to 96-well plates at a seeding rate of 5 × 10³ cells per well and permitted 24 h to settle and adhere. Post-adhesion, these cells received treatments with variable concentrations of DSF, CuET, dsMN, dsMCu, dsMCu-D, dsMCu-D@M, and dsMCu-D@M-Co for a duration of 12 h. Subsequent to the treatment period, 20 µL of MTT reagent (5 mg/mL) was added to each well, followed by a further incubation of four hours. After this incubation, the medium was evacuated, and 50 µL of DMSO was introduced to each well to solubilize the formazan deposits, involving gentle agitation for 15 min. The absorbance of each well was then quantified at 570 nm using a Bio-Rad Model 680 Microplate reader. To assess cellular viability, cells in 12-well plates were incubated for 12 h, then stained with Calcein-AM and PI for 25 min at 37 °C, and observed under an inverted fluorescence microscope after the treatment. For apoptosis studies, BxPC-3 cells were arranged in 12-well plates and exposed to PBS, dsMCu, dsMCu-D, dsMCu-D@M, and dsMCu-D@M-Co (50 µg/mL) for 12 h. The cells were then released using non-EDTA trypsin, collected, and subjected to Annexin V-FITC/PI staining, followed by flow cytometry analysis to measure apoptotic activity.
Nanoparticle penetration in vitro
The 96-well plates were coated with 1.5% agarose solution, which had been heated to a high temperature. The setup was incubated at 37 °C for 24 h. The BxPC-3 cells were seeded into the agarose-coated wells at a concentration of 1 × 10³ cell/well. After seven days of incubation, BxPC-3 MCSs were developed and those ranging in size to ~ 200 μm in diameter were chosen for continued testing. To test the ability of the nano platform to penetrate, the prepared BxPC-3 MCS was placed into a confocal dish with 1 mL of the new medium; this was about a new medium that had been labelled with FITC forms of dsMCu-D, dsMCu-D@M, and dsMCu-D@M-Co. The CLSM technique was employed to perform Z-stack scanning at every five µm from the top to bottom of the MCS after 24 h of incubation. The MCS was dissociated into single cells with accurate reagent, and FITC-positive cells and fluorescence intensity were measured using flow cytometry. In the study of MCS growth inhibition, the prepared BxPC-3 MCSs were co-incubated with dsMCu, dsMCu-D, dsMCu-D@M, and dsMCu-D@M-Co at a concentration of 50 µg/ml. The MCSs remained in culture for five days, during which their development was traced and recorded based on photographs taken by an optical microscope.
Assessment of nanoparticles triggered DLAT aggregation by CLSM
The BxPC-3 cells were seeded and cultured in an 8-chamber slide for 24 h. Subsequently, five treatments were administered: PBS, dsMCu, dsMCu-D, dsMCu-D@M, and dsMCu-D@M-Co. Following this, the cells were washed with cold PBS and fixed with 4% paraformaldehyde for 30 min. Subsequently, Triton X-100 was applied for 10 min to ensure proper permeabilization. After blocking with a blocking buffer for 2 h, the primary antibody to DLAT was incubated with the cells overnight at 4 °C. Following a PBS wash, the cells were incubated with the corresponding secondary antibody for 2 h at room temperature. DAPI and actin-tracker were utilized to label the nucleus and F-actin, respectively. Finally, fluorescent images were captured using CLSM.
Western blot analysis
Cell cultures were separated, using different treatments at a temperature of 37 °C. Following a 12-hour incubation period, the cells were washed extensively using PBS at low temperature and subsequently subjected to lysis using RIPA buffer for lysis. These lysed samples were centrifuged at 12,000 rpm at 4 °C for 15 min. 20 µg of protein in each lane was isolated through 10% SDS-PAGE. The isolated proteins were moved onto a polyvinylidene fluoride (PVDF) membrane and obstructed using 5% skim milk powder for 2 h at ambient temperature. Subsequently, this membrane was subjected to overnight incubation with primary antibodies including DLAT, LIAS, NPLOC4 and β-actin. Subsequently, this membrane was additionally exposed to the HRP-labeled goat anti-rabbit secondary antibody for 2 h at a temperature of 4 °C. An image was taken using a molecular imager (ChemiDoc Touch Imaging System, Bio-Rad, USA).
Detection of mitochondrial membrane potential and ROS production
BxPC-3 cells were cultured in an 8-chamber slide at 37 °C for 24 h prior to applying various treatments. Eventually, the cells were stained with the JC-1 probe and imaged using CLSM.BxPC3 cells were seeded into 8-chamber slide and cultured at 37 °C for 24 h. Afterwards, cells were incubated with PBS, dsMCu, dsMCu-D, dsMCu-D@M, and dsMCu-D@M-Co. Cells were washed with PBS after endocytosis and incubated with DCFH-DA for 20 min at 37 °C. The intracellular oxidized DCF could be used as the indicator of ROS generation. The corresponding fluorescence images of intracellular DCF at excitation wavelength of 488 nm were taken by CLSM. Moreover, the cells could be collected and analyzed by flow cytometry in addition to FCM observation.
Blood compatibility test
2 mL whole mouse blood was taken from the BALB/c nude mice and added into the anticoagulant tube. After centrifugation (3000 rpm, 5 min) and washing the sediment with PBS five times, the red blood cells were collected and then diluted with PBS three times. 100 µL of the above diluted red blood cells were added to tubes containing 900 µL of dsMCu, dsMCu-D, dsMCu-D@M, and dsMCu-D@M-Co solution, ultrapure water, or PBS and mixed evenly. After incubation at 37 °C for 2 h, the mixtures were centrifuged at 10,000 rpm for 5 min, and the UV absorbance of each tube at 540 nm was measured.
In vivo biodistribution
BxPC-3 tumor-bearing mice (n = 3 per group) were randomly assigned to receive intravenous injections of Cy5.5-labeled dsMCu-D, dsMCu-D@M, or dsMCu-D@M-Co (10 mg/kg). The mice underwent in vivo imaging using the IVIS Spectrum fluorescence imaging system (Caliper, USA) at 12-, 24-, and 48-hours post-injection. At the final time point, tumors and major organs (heart, liver, spleen, lung, and kidneys) were collected for ex vivo biodistribution analysis. Copper content was quantified using ICP-MS.
In vitro and in vivo MRI
dsMCu-D@M-Co nanoparticles were dispersed in deionized water at copper ion concentrations ranging from 0 to 1.6 mM and then aliquoted into 1 mL Eppendorf tubes for analysis. T1-weighted images were acquired using a 3T MRI scanner (Discovery MR 750, GE, USA). For in vivo studies, the nanoparticles were administered intravenously to mice through the tail vein. Subsequent imaging was performed using a rapid spin-echo sequence with a repetition time of 400 ms, an echo time of 21 ms, a field of view of 60 × 60 mm, a matrix of 320 × 192, and a slice thickness of 2 mm.
In vivo assessment of collagen degradation and tumor penetration
To evaluate collagen degradation within tumor tissues, Masson’s trichrome staining and immunofluorescence methods were employed. BxPC-3 tumor-bearing mice were euthanized 24 h post-administration of dsMCu, dsMCu-D, dsMCu-D@M, or dsMCu-D@M-Co (10 mg/kg). Tumor samples were processed into paraffin-embedded sections, were Masson’s trichrome staining highlighted collagen for microscopic analysis. Furthermore, these sections underwent incubation with primary antibodies against collagen I, followed by secondary antibodies as per the staining kit’s protocol. To evaluate the in vivo penetration ability of dsMCu-D@M-Co, mice bearing BxPC-3 tumors were administered intravenous injections of FITC-labeled dsMCu-D, dsMCu-D@M, and dsMCu-D@M-Co at a dosage of 10 mg/kg. Forty-eight hours post-injection, the mice were euthanized, and their tumors were promptly excised, snap-frozen, and sectioned. The sections of these tumors were then stained with DAPI and CD31 to assess penetration dynamics, which were examined using confocal laser scanning microscopy (CLSM).
In vivo antitumor effect
When the tumor reaches the desired volume, BxPC-3 tumor-bearing mice (n = 5 per group) were randomly assigned to receive treatments including PBS, dsMCu, dsMCu-D, dsMCu-D@M, and dsMCu-D@M-Co. Injections were administered on days 1, 3, and 5. Tumor volume and body weight were recorded every two days. On day 14 post-treatment, the tumors were excised, and the survival of mice in each group was monitored for up to 60 days.
IHC and IF staining analysis
In the case of IHC staining, after dewaxing, sections underwent antigen retrieval in citrate sodium buffer at 60 °C overnight. Following this, sections were exposed to 3% H2O2 and obstructed using 5% bovine serum albumin (BSA) at room temperature for 1 h. The sections were incubated overnight with the corresponding primary antibodies at 4 °C. Subsequently, they were incubated with biotin-labeled secondary antibodies at room temperature for 1 h. The DAB reagent was then applied, and images for IHC were captured using a microscope. In the case of IF staining, dewaxing and antigen retrieval procedures mirrored those used in IHC staining. Following this, the sections were subjected to permeabilization using 0.1% Triton X-100 for 20 min and obstructed using 5% BSA for 1 h at room temperature. After a PBS wash, the sections were exposed to the corresponding secondary antibodies and stained with DAPI. Images for IF were obtained using a fluorescence microscope.
Statistical analysis
GraphPad Prism software was employed for statistical analysis. The findings were expressed as mean ± standard deviation (SD). Group comparisons were evaluated using both one-way analysis of variance (ANOVA) and Student’s t-test, with statistical significance set at P < 0.05. Significance levels were denoted as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.





留言 (0)