Nanoparticle characterization
Different coated (polyvinylpyrrolidone (PVP; nanoComposix, USA), citrate (nanoComposix, USA), and polyethylene glycol (PEG; nanoComposix, USA)) and sized (5 nm and 50 nm) silver (Ag) NPs were used. Dynamic light scattering (DLS) and zeta potential (ZP, mV) of Ag-NPs were measured using the Zetasizer Nano ZS (Malvern, instruments, UK). Ag-NP concentration and size was detected using NanoSight LM10 (Malvern Instruments, Worcestershire, UK). Imaging of Ag-NPs was performed using high resolution transmission electron microscopy (TEM 1400, Jeol, Philips, NL) at magnifications: X8k (500 nm Ag-NP), X50k (100 nm Ag-NP), X150k (20 nm Ag-NP).
NP degradation assay
Silver citrate 5nm NPs used in the following experiments were assessed for their degradation in media at varying PH levels (7.4 and 4.5). For this end, Ag-citrate-5nm NPs were incubated at 5, 10, 15, 20, and 25 µg/ml concentrations in cell culture media adjusted to the various acidity levels. Free Ag ion detection was performed at ambient temperature using 3,3’,5,5’,-tetramethylbenzidine (TMB from Sigma-Aldrich, Belgium) as a highly sensitive and selective colorimetric indicator in NaAc buffer (pH: 4.0). Absorbance changes in each well containing the Ag-citrate-5 nm NPs were measured at 656 nm according to the manufacturer’s instructions. In a standard procedure, 200µL of TMB solution (1 mM in ethanol) was introduced to 800 µL of NaAc buffer. After allowing the reaction to proceed for 30 min at room temperature, UV–vis spectra were obtained using a plate reader.
Cell culture
Mouse breast adenocarcinoma (4T1), mouse renal adenocarcinoma (Renca), mouse colorectal carcinoma (CT26), human cervical cancer (HeLa), human lung cancer (A549), and mouse mesenchymal stem (mMSC) cell lines were used. All cell lines were obtained commercially (Atcc, Belgium). 4T1, HeLa and A549 cells were cultivated in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Thermo Fisher Scientific, Belgium) supplemented with 10% fetal bovine serum (FBS; Gibco, Thermo Fisher Scientific, Belgium) and 1% penicillin/streptomycin (Corning, Belgium). Renca cells were cultivated in Roswell Park Memorial Institute medium (RPMI 1640 – GlutaMaxTM; Gibco, Thermo Fisher Scientific, Belgium) supplemented with 10% FBS, 1mM sodium pyruvate (Gibco, Thermo Fisher Scientific, Belgium), 1% non-essential amino acids (HyClone, Fisher Scientific, Belgium) and 1% penicillin/streptomycin (Croning, Belgium). CT26 cells were cultivated in RPMI 1640 supplemented with 10% FBS, and 1% P/S. mMSC were cultivated in DMEM supplemented with 10% FBS and 10% horse serum (HS; Gibco, Thermo Fisher Scientific, Belgium). All cell types were incubated at 37 °C in a 5% CO2 humidified environment.
In vitro nanoparticle cytotoxicity evaluation
To analyze nanoparticle cytotoxicity, cells were seeded during their exponential growth phase. A549, Renca, CT26, HeLa, and mMSC were seeded at a density of 2000 cells/well, 4T1cells were seeded at a density of 1500 cells/well in sterile 96-well plates (Costar, Belgium) and incubated for 24 h at 37 °C and at 5% CO2. The next day, cells were treated with increasing concentrations (0, 1 5, 10, 15, 20, 25 µg/ml) of inorganic NPs. Different NP coatings were assessed: polyvinylpyrrolidone (PVP; nanoComposix, USA), citrate (nanoComposix, USA), and polyethylene glycol (PEG; nanoComposix, USA). 4%Fe-Cu, 2%Fe-ZnO, 33%Cu-TiO2 were a part of a collaboration. Serial dilutions of NPs in total cell culture medium (according to the cell type) were prepared per type of differently coated Ag NPs and administered in triplicates. After 24 h of incubation, the cells were washed twice with Phosphate Buffered Saline (PBS; Gibco, Thermo Fisher Scientific, Belgium), followed by staining for further microscopic analysis.
Analysis of viability and mitochondrial oxidative stress
Following exposure to the NPs, cells were washed twice with PBS and incubated for 30 min with 100 nM Image-IT DEAD green viability stain (Thermo Fisher Scientific, Belgium) and 200 nM Mito Tracker RED CMXRos (Thermo Fisher Scientific, Belgium) in total cell culture medium (100 µL/well). Cells were then washed with PBS and fixed in 2% paraformaldehyde (PFA) (pH = 7.4) for 10 min at room temperature (RT). After fixation, the cells were counterstained with 0,2% Hoechst nuclear stain (Hoechst 33342; Thermo Fisher Scientific, Belgium) in PBS for 10 min. Viability, mitochondrial stress and the generation of reactive oxygen species (ROS) were measured using an automated epifluorescence microscope (INCell Analyzer 2000, GE Healthcare Life Science; Light Microscopy and Imaging Network (LiMoNe), VIB-KU Leuven). The microscope was set to detect FITC (viability), dsRed (mitochondrial stress and oxidative stress) and DAPI (cell nucleus). Twelve random image fields were obtained per well with a 20X objective (NA 0.45). Analysis of fluorescent images was performed using the IN Cell Investigator software (GE Healthcare Life Science). Cell nuclei (DAPI) were first identified and segmented, after which FITC intensity signals and mitochondrial intensity signals were determined and linked, via segmentation, to the cell nucleus of the cell to which they belonged, after which the intensity and the area were determined per cell. Cell viability was then determined as: the number of cells with FITC signal at the cell nucleus; mitochondrial ROS was determined as: the intensity of the mitochondrial network of the cell, and mitochondrial stress as: the total area of the mitochondrial network of the cell. These values were always normalized to the control cells included in each assay to avoid inter-variability between different experiments. Viability, ROS and mitochondrial stress are therefore expressed as percent relative values.
Analysis of cellular morphological changes
After incubation with the different NP formula’s, cells were washed twice with PBS and fixed for 10 min with 2% PFA (pH = 7.4) at RT. After fixation, cells were incubated for 1 h with Actin-stainTM 488 phalloidin (Cytosceleton, Inc, Belgium). After staining, the cells were washed 2 x with PBS and counterstained for 10 min with 0,2% Hoechst nuclear stain. The actin area was measured using an automated epifluorescence microscope (Operetta CLS, PerkinElmer; Light Microscopy and Imaging Network (LiMoNe), VIB- KU Leuven). The microscope was set to detect FITC (actin area), and DAPI (cell nucleus). Twelve Random image fields were obtained per well with a 20X objective (NA 0.45). Analysis was performed using the Cell Profiler cell analysis software (Broad Institute, USA). The different cell nuclei were first identified and segmented, after which the FITC signal was determined by means of intensity values. Cell actin area was then determined as the total area of the actin network of the cell, stained with Actin-stain TM 488-Phalloidin. These values were always normalized to the control cells included in each assay to avoid inter-variability between different experiments.
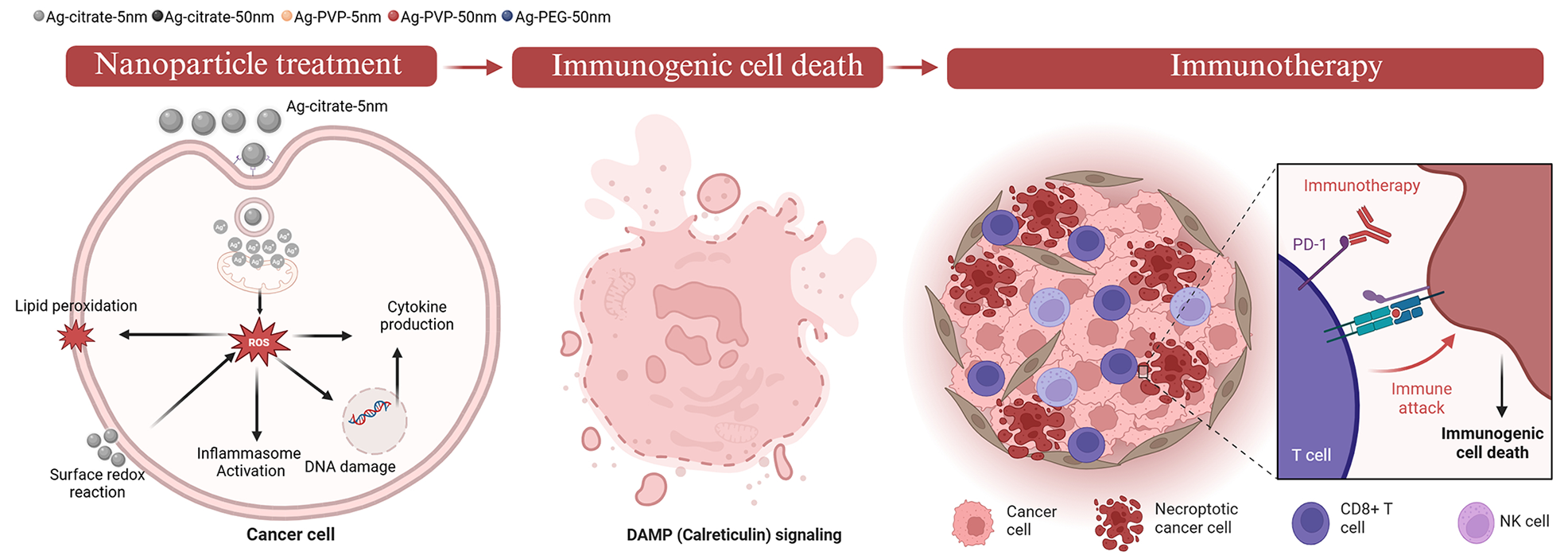
Analysis of immunogenic cell death
Renca cells were seeded in a 12-well plate (Greiner Bio-one, Belgium) with a density of 1,2 × 105 cells/well. Renca cells were incubated with Ag-citrate-5nm NPs at a concentration of 5 µg/ml for 24 h. Supernatant was discarded and cells were washed 1 time with 1mL PBS. 500µL of 0.25% trypsin (Gibco™ Trypsin-EDTA (0.25%), phenol red, fisher scientific, USA) was added to the cells and incubated for 5 minutes at 37 °C. 1 mL of cell media was added directly in the wells and mixed, media and cells were then transferred to test tubes. The tubes were centrifuged at 400 x g for 5 min and the supernatant was discarded. The cells were washed 1 time with 1 x PBS and centrifuged again at 400 x g for 5 min, the supernatant discarded, and the cells were stained with recombinant anti-calreticulin antibody (Recombinant Alexa Fluor® 647 Anti-Calreticulin antibody- ER Marker (ab196159), abcam, UK) and MHC class I antibody (MHC Class I (H-2Kd/H-2Dd) Monoclonal Antibody (34-1-2 S), FITC, eBioscience™, Thermo Fisher science, Belgium) for 1 h on ice. Cells were washed with 2 mL washing solution (WS; 1 x PBS + 1%FBS) and centrifuged at 400 x g for 5min. Hereafter, cells were washed with 1 x PBS and centrifuged at 400 x g for 5 min. Cells were re-suspended in 1xPBS and transferred to 1,5 mL micro test tubes (total volume not more than 200 µL) and detected using image-based flow cytometry (Amnis® ImageStream®X Mk II, Luminex, USA). Analysis was performed using IDEAS application v6.0 (Luminex, USA).
Analysis of cytokine expression
Renca cells were seeded in a 12-well plate (Greiner Bio-one, Belgium) with a density of 1,2 × 105 cells/well. Renca cells were incubated with Ag-citrate-5nm NPs at a concentration of 2 µg/ml and 5 µg/ml for 24 h. Supernatant was discarded and cells were washed 1 time with 1mL PBS. The cells were incubated with brefeldin A (3 µg/ml) (Sigma-Aldrich, USA)(3 µg/mL) for 4 h at 37 °C. After incubation the cells were washed with 1 x PBS and 500 µL of 0.25% trypsin (Gibco™ Trypsin-EDTA (0.25%), phenol red, fisher scientific, USA) was added to the cells and incubated for 5 minutes at 37 °C. 1mL of cell media was added directly in the wells and mixed, media and cells were then transferred to test tubes. The tubes were centrifuged at 400 x g for 5 min and the supernatant was discarded. The cells were washed 1 time with 1x PBS and centrifuged again at 400 x g for 5 min, the supernatant discarded. The cells were fixed for 10 min with 2% PFA (pH = 7.4) at RT. The cells were washed 1 time with 1 x PBS and centrifuged again at 400 x g for 5 min, the supernatant discarded. Permeabilization was done using Trition 100x (final concentration 1 x in PBS) for 10 min at RT. The cells were washed again with 1 x PBS at 400 x g for 5 min. The cells were incubated with Fc receptor blocking antibody (CD16/CD32 Rat anti-Mouse, Unlabeled, Clone: 2.4G2, BD, fisher scientific) in washing solution (WS; 1 x PBS + 1% FBS) for 30 min on ice. After which 2 mL of WS was added and centrifuged at 400 x g for 5 min. The supernatant was discarded and the cells were incubated with primary antibodies (anti- IFN-alpha/beta R2 PE-conjugated (Mouse IFN-alpha / beta R2 PE-conjugated Antibody (FAB1083P), PE, Biotechne), anti-Interferon gamma violetFluor™ 450 (Anti-Interferon gamma antibody (ab253083), violetFluor™ 450, abcam), anti-IL-6 FITC (IL-6 Monoclonal Antibody (MP5-20F3), FITC, eBioscience™, Thermo Fisher), anti-IL-12 p35 (IL-12 p35 Monoclonal Antibody (4D10p35), eFluor™ 660, eBioscience™, Thermo Fisher); and TNFα (TNF alpha Monoclonal Antibody (MP6-XT22), PE-Cyanine7, eBioscience™, Thermo Fisher). for 1 h on ice (protected from light). After incubation, 2 mL of WS was added and centrifuged at 400 x g for 5 min. Cells were then washed with 1 x PBS and re-suspended with 1 x PBS and transferred to 1,5 mL micro test tubes (total volume no more than 200 µL). Antibody-Labelled cells were detected using image-based flow cytometry (Amnis® ImageStream®X Mk II, Luminex, US). Analysis was performed using IDEAS application v6.0 (Luminex, US).
Animal experiment and ethic
Animals were kept in filter top cages with controlled temperature (21 ± 2 °C), humidity (50 ± 10%) and day-night cycle of 12 h/12 h. Mice received ad libitum standard pellet diet, and water. Mice were followed up daily. All animal experiments were approved by the ethical commission of animal experiments (ECD) of KU Leuven (approval number: P218/2018 and P219/2018) and were in accordance with principles and procedures in national and European regulations.
Renca subcutaneous tumor model
Renca cells were transduced with a viral vector containing the firefly luciferase gene. Firefly luciferase uses luciferin as a substrate and emits light at 560 nm that was detected via non-invasive bioluminescence optical imaging. In this study, 51 female BALB/c mice were used (5 weeks old) (Charles river, Beerse, Brussel). Mice were injected subcutaneously, at the bottom of the right flank, with 1,0 × 106 RencaLuc + cells.
In vivo bioluminescence imaging (BLI)
Tumor growth was monitored twice a week, using a non-invasive bioluminescence (BLI) optical imaging system (IVIS spectrum; PerkinElmer). For each session, mice were injected intraperitoneally (IP) with 20 mM Luciferin (MedChem Express, USA). The mice were then anesthetized and positioned in the IVIS Spectrum. BLI images were obtained 10 min post-administration of Luciferin (medium binning, f stop = 1, excitation time = 20 s) under general anesthesia with 2% isoflurane inhalation. Regions of interest (ROI) were indicated covering the bioluminescence signal from the tumor. Images were analyzed with LIVING Image processing software (Perkin Elmer, Waltham, MA).
In vivo elastase – caspase fluorescence imaging
At the end of the experiment, mice were anesthetized and injected intravenously with Neutrophil Elastase 680 FAST (Neutrophil Elastase™ 680 FAST (NEV11169), Waltham, MA) and NIR-FLIVO 747 Tracer (NIR-FLIVO 747 Tracer In vivo Assay, Immunochemistry Technologies). The mice were then positioned in the IVIS Spectrum. Fluorescence images were obtained 4 h post-administration of the tracers (excitation/emission Elastase: 675 nm/720nm, excitation/emission NIR-FLIVO: 747 nm/776nm) under general anesthesia with 2% isoflurane inhalation. Regions of interest (ROI) were indicated covering the fluorescence signal from the tumor. Images were analyzed with LIVING Image processing software (Perkin Elmer, Waltham, MA).
Combination treatment protocol
After 14 days, tumor size was measured in Renca flank tumor bearing mice using non-invasive BLI. 51 mice in total were divided into 9 groups: group 1 = control group (PBS, n = 9); group 2 = NP-treatment (n = 8); group 3 = immunotherapy anti-PD1 (IT, n = 7); group 4 = combination therapy where only the IT was boosted (NP + IT(boost), n = 7), group 5 = combination therapy where the NP and the IT were boosted (NP (boost) + IT (boost), n = 5), group 6 = anti-CD8 (n = 4), group 7 = NP + anti-CD8 (n = 4), group 8 = anti-PD1 + anti-CD8 (n = 4), group 9 = NP + anti-PD1 + anti-CD8 (n = 4). Immunotherapy was based on monoclonal antibodies against immune checkpoint PD-1 (CD279, InVivo MAb anti-mouse PD-1, Biocell). Anti-CD8 was based on monoclonal antibody against CD8 alpha (In vivo MAb anti-mouse CD8α, Biocell). Here, mice from groups 3,4,6,7,8 and 9 (anti-PD1, anti-CD8 or both), received intraperitoneal booster injections on day 18, 22 and 26 of 150 µg/mice. NP treatment was based on peritumoral injection of Ag-citrate 5 nm NPs on day 14 with a concentration of 20 µg/mouse under general anesthesia with 2% isoflurane inhalation. Mice from group 5 received peritumoral booster injection of Ag-citrate-5 nm and intraperitoneal booster injection of anti-PD1 on day 18, 22, and 26. Control animals were injected peritumorally with PBS on day 14. Tumor growth was monitored twice a week. All mice were euthanized after 30 days from tumor cell injection.
Another cohort of Renca flank tumor bearing mice were treated with higher concentration of Ag-citrate-5 nm (50 µg/mouse) and anti-PD1 (200 µg/mouse). 32 mice were divided into 4 groups: group A = control (PBS, n = 8), group B = NP- treatment (n = 8), group C = anti-PD1 group (n = 8), and group D = NP + anti-PD1 (n = 8). All the groups received booster injections on day 9, 12, 15, and 18 under general anesthesia with 2% isoflurane inhalation.
Tissue preparation
Vital organs and tumors were fixed in 4% PFA (PFA, clinipath, VWR, Belgium). First, organs/tumors were incubated in 15% sucrose (Sigma-Aldrich, USA) (in 1 x PBS) overnight at 4 °C. The next day they were incubated in 30% sucrose overnight at 4 °C, after which they were embedded in embedding molds (Peel-A-Way embedding molds, Sigma-Aldrich, USA) using O.C.T (Tissue-Tek, Sakura Finetek, USA). Sections of 10 μm were processed using a cryostat (CryoStar NX70, Thermo Fisher Scientific) and fixed on microscope slides (Super Frost Plus, VWR, Belgium).
Splenocyte isolation and staining
Fresh spleen was collected in 3 mL complete medium (CM; DMEM + 10%FBS + 1% penicillin/streptomycin) in a homogenization tube (gentleMACS™ C Tubes, Miltenyi Biotec). Spleen was dissociated using tissue dissociator gentle MACS Octo (gentleMACS™, Miltenyi Biotec). The cell suspension was transferred to a 70 μm Nylon strainer (greiner bio-one, Belgium). The suspension was collected in a 50 mL tube, transferred to a 15mL tube and centrifuged at 400 x g for 5 min. After centrifugation, the supernatant was discarded, and the pellet was re-suspended in 3 mL red blood cells lysis buffer (NH4Cl). Blood lysis was stopped by adding 10 mL of CM and then centrifuged at 400xg for 5 min. The cells were washed 2 times with 1 x PBS and the pellet was re-suspended with PBS and transferred to test tubes: 1/3 of each control sample was taken, mixed and split in 8 test tubes (single antibody staining for matrix compensation) -the remaining cells (2/3) or each control samples were split in 2 tubes (for cocktail A and B antibodies). Cell samples derived from treated spleens were split in 2 test tubes (for cocktail A and cocktail B). The tubes were centrifuged at 400 x g for 5 min and the supernatant was discarded. The cells were incubated with Fc receptor blocking antibody (CD16/CD32 Rat anti-Mouse, Unlabeled, Clone: 2.4G2, BD, fisher scientific) in washing solution (WS;1xPBS + 1%FBS) for 30 min on ice. After which 2 mL of WS was added and centrifuged at 400 x g for 5 min. The supernatant was discarded and the cells were incubated with primary antibodies (cocktail A: anti-CD3 eFluor 450 (CD3e Monoclonal Antibody (145-2C11), eFluor 450, eBioscience™, Thermo Fisher), anti-CD19 PE (CD19 Monoclonal Antibody (MB19-1), PE, eBioscience™, Thermo Fisher), anti-F4/80 FITC (F4/80 Monoclonal Antibody (BM8), FITC, eBioscience™, Thermo Fisher), anti-CD45 PE-Texas Red (CD45 Monoclonal Antibody (30-F11), PE-Texas Red, Thermo Fisher); and cocktail B: anti-CD4 APC (CD4 Monoclonal Antibody (GK1.5), APC, eBioscience™, ThermoFisher), anti-CD8α PE-Texas Red (CD8 alpha Monoclonal Antibody (5H10), PE-Texas Red, Thermo Fisher), anti-CD69 PE (CD69 Monoclonal Antibody (H1.2F3), PE, eBioscience™, Thermo Fisher), anti-CD38 eFluor450 (CD38 Monoclonal Antibody (90), eFluor 450, eBioscience™, Thermo Fisher)) for 1 h on ice (protected from light). For the matrix compensation, cells from control group were stained separately with the single antibodies. After incubation, 2 mL of WS was added and centrifuged at 400 x g for 5 min. Cells were then washed with 1 x PBS and re-suspended with 1xPBS and transferred to 1,5 mL micro test tubes (total volume no more than 200 µL). Antibody-Labelled cells were detected using image-based flow cytometry (Amnis® ImageStream®X Mk II, Luminex, US). Analysis was performed using IDEAS application v6.0 (Luminex, US).
Immunohistochemistry
10-µm cryostat sections were prepared for immunohistochemical staining. Antigen retrieval was achieved by incubation with proteinase K for 15 min. Sections were washed with 1 x PBS for 5 min and incubated with blocking solution (1 x PBS + 10% normal goat serum (NGS, Thermo Fisher scientific) + 1% FBS) for 1 h. The sections were washed 2 times with 1 x PBS and incubated in avidin blocking solution (0.001% avidin (Sigma-Aldrich, USA) in 1 x PBS) for 20 min. The sections were washed twice with 1xPBS and incubated with biotin blocking solution (0.001% biotin (Sigma-Aldrich, USA) in 1xPBS) for 20 min. After washing twice with 1xPBS, sections were incubated overnight at 4 °C with anti-F4/80 antibody (F4/80 antibody | Cl: A3-1, Bio-Rad, USA). Endogenous peroxidases were quenched by incubating the sections in 3% hydrogen peroxide (Acros Organics, Thermo Fisher Scientific, Belgium) for 20 min. The sections were then washed twice with 1xPBS. Incubation with Anti-Rat-Biotin (Biotin-SP Affini Pure Goat Anti-Rat IgG (H + L), Jackson Immuno research, UK) was performed for 1 h at RT. After washing twice in 1 x PBS, the sections were incubated with streptavidin-HRP (HRP-Conjugated Streptavidin, Thermo Fisher) for 30 min at RT. After washing with 1 x PBS, the sections were incubated with Tyramide working solution (Alexa Fluor™ 488 Tyramide SuperBoost™ Kit, goat anti-rabbit IgG, Invitrogen, Thermo Fisher scientific) by following the manufacturer instructions. Stop reagent (Alexa Fluor™ 488 Tyramide SuperBoost™ Kit, goat anti-rabbit IgG, Invitrogen, Thermo Fisher scientific) was added to each section for 2 min after which the sections were washed 3 times with 1 x PBS. Sections were incubated overnight with anti-CD8 antibody (Novus Biologicals, UK). The sections were washed twice with 1xPBS and incubated with goat-anti-rabbit-polyHRP (Invitroge, Thermo Fisher) for 1 h at RT. After incubation, the sections were washed once with 1 x PBS and incubated for 1 h with Tyramide working solution (Alexa Fluor™ 594 Tyramide SuperBoost™ Kit, goat anti-rabbit IgG, Invitrogen, ThermoFisher scientific) by following the manufacturer instructions. Stop reagent (Alexa Fluor™ 594 Tyramide SuperBoost™ Kit, goat anti-rabbit IgG, Invitrogen, Thermo Fisher scientific) was added for 2 min and the sections were washed 3 times with 1 x PBS. Hoechst staining was added for 10 min at RT and the sections were washed 2 times with 1xPBS. Mounting was performed using fluoro-mount aqueous mounting medium (Sigma-Aldrich, USA) and the sections were covered using cover glass (Rectangular cover glasses, VWR, Belgium). The images were acquired with Vectra Polaris automated system (Akoya Biosciences, USA) and analyzed using open-source software QuPath.
Histology
Eosin-Hematoxylin staining − 10 μm cryostat sections were brought to RT for 30 min in Mili-Q water. Tissue sections were washed for 5 min in 1 x PBS and stained with hematoxylin (Sigma-Aldrich, USA) for 3 min (protected from light). Hereafter, slices were washed with Mili-Q water for 5 min and washed with 80% EtOH (VWR, Belgium) – 0.15% HCl (Acros Organics, Thermofisher Scientific, Belgium) solution for 1 min. Slides were washed again with Mili-Q water for 1 min whereafter they were put in ammonia water (0.3% v/v) for 30 s. After washing with Mili-Q water for 5 min, they were washed with 95% ethanol for 1 min and stained with eosin (Sigma-Aldrich, USA) for 1 min (protected from light). After staining, dehydration was done by washing with 95% ethanol for 5 min followed by two washings with 100% ethanol (5 min each time). After dehydration, clearing of the slides is performed with 100% xylene (clinipath, VWR, Belgium) (5 min each). Mounting was done with DPX (Merck KGaA, Germany) and cover glass (Rectangular cover glasses, VWR, Belgium). Images of tissue sections were acquired with the automated Vectra Polaris system (Akoya Biosciences, USA) and analyzed with the open-source software FIJI.
Statistics
Statistical analysis was performed using GraphPad Prism 8.0 (GraphPad Software, USA). Data were presented as mean ± standard error to the mean (SEM). Statistical comparisons between different groups were analyzed using two -way Anova and one-way Anova with the application of Bonferroni correction. The level of statistical significance was indicated when p < 0.05 (*: p < 0.05; **:p < 0.01; ***:p < 0.001, ****:p < 0.0001).





留言 (0)