
記住我


Chronic inflammatory diseases affect more than 41 million individuals annually worldwide, with more than 17 million deaths occurring before the age of 70 [1]. Notably, over 77% of reported deaths from noncommunicable diseases are concentrated in low-income or middle-income countries [2]. For example, India, as the second most populous country globally, reports that 63% of deaths are attributed primarily to chronic diseases [3], such as cardiovascular diseases (27%), chronic respiratory diseases (11%), cancer (9%), and diabetes (3%) [4]. The significant impact of these conditions highlights a critical public health challenge, emphasizing the need to develop new therapeutic strategies to manage and mitigate the effects of chronic inflammatory diseases.
Inflammation is a central biological process in several diseases. This may be triggered by components of the bacterial wall and other pathogen-associated molecular pattern products [5, 6]. These include lipopolysaccharides (LPS), formyl methionyl peptides, lipopeptides, peptidoglycans, flagellins, and microbial DNA/RNA that serve as noxious stimuli [7]. Recognized by Toll-like receptors (TLRs), nucleotide-binding and oligomerization domain-containing receptors [NOD-like receptors (NLRs)], and retinoic acid-inducible genes [RIG I-like receptors (RLRs)], these molecules activate downstream signaling pathways through nonreceptor protein kinases [8, 9]. This culminates in the activation of transcription factor networks such as nuclear factor κB (NF-κB), activation protein 1 (AP-1), interferon regulatory factors (IRFs), and signal transducers and activators of transcription (STATs), which are crucial for regulating gene expression during inflammatory responses [10]. Persistent immune activation, which contributes to chronic inflammation, underlies the progression of diverse diseases, including atherosclerosis, cancer, asthma, rheumatoid arthritis, fatty liver syndrome, inflammatory bowel disease, Alzheimer's disease, and type I diabetes [11]. Pathophysiologically, chronic inflammation is driven by immune cells such as macrophages, T cells, and B cells, which release proinflammatory mediators such as cytokines and chemokines, maintaining a cycle of inflammation [12]. The prognosis of chronic inflammation depends on the underlying cause and the effectiveness of management strategies. Cellular and molecular biomarkers, including reactive oxygen species (ROS), reactive nitrogen oxide species (RONS), cytokines, C-reactive proteins, and prostaglandins, are closely associated with inflammatory processes and serve as prognostic indicators [13]. For example, elevated ROS levels produced by activated macrophages in the tumor microenvironment promote tumor growth and metastasis [14]. This sustained inflammatory response not only reduces quality of life by prolonging symptoms and impairing functionality but also poses risks of tissue damage and promotes organ dysfunction [6]. The development of therapeutics targeting chronic inflammation is crucial for alleviating immediate symptoms, preventing long-term complications, and enhancing overall health outcomes. Moreover, addressing chronic inflammation has broader implications for reducing the economic burden on healthcare systems and promoting global public health.
Delivering high-quality support, services, and information plays a crucial role in empowering patients to manage chronic inflammatory conditions, thus forming a key aspect of treatment [15]. However, common anti-inflammatory drugs may pose risks. Disease-modifying antirheumatic drugs (DMARDs) require monitoring for infections and liver damage; glucocorticoids may lead to resistance, and TNF-blockers can worsen multiple sclerosis (MS) symptoms [16,17,18,19]. Few drugs have both anti-inflammatory and pro-reparative effects, and most clinical trials have focused on reducing inflammatory mediator levels rather than enhancing tissue repair. Consequently, there are currently no FDA-approved drugs that target inflammation and lead to tissue repair. Fortunately, the development of targeted therapeutics that can address a spectrum of chronic inflammatory conditions is underway, and several randomized clinical trials have focused primarily on individuals with specific chronic inflammatory diseases. Several candidate therapies have advanced to phase II and III trials on the basis of the optimistic outcomes of randomized control trials [20]. This underscores the complexity of and ongoing efforts to develop effective and comprehensive treatments for chronic inflammatory diseases.
Given the extended period required for novel therapeutic compounds to come to the clinic, the concept of drug repurposing—finding new therapeutic implications for already established compounds—has become valuable [21]. These candidate compounds offer several advantages; namely, pharmacodynamic and pharmacokinetic studies have already been performed, toxicology studies have been performed, and chemical optimization has been complete, leading to significant development time and cost reductions [22]. Drug repurposing, characterized by investigating new indications for previously approved drugs or advancing previously studied but unapproved drugs, is a fundamental approach in drug development. Approximately 30–40% of new drugs and biologics approved by the U.S. Food and Drug Administration (FDA) between 2007 and 2009 can be categorized as repurposed or repositioned products [23]. Indeed, repurposed drugs have already undergone extensive preclinical and clinical testing for their original indications, saving significant time and resources [24] and having well-established safety profiles, reducing the likelihood of unexpected adverse effects in clinical trials [25]. Moreover, repurposing leverages existing knowledge regarding drug mechanisms of action and pharmacokinetics, facilitating faster translation from bench to bedside [26]. Overall, repurposing offers a streamlined approach for drug development, potentially accelerating the availability of effective treatments for chronic inflammatory diseases. The concept of drug repurposing has gained significant attention during the COVID-19 pandemic, particularly as the FDA granted emergency authorization for the use of several drugs to treat COVID-19. For example, within six months of the onset of the pandemic, remdesivir, which was originally developed for RNA-based viruses, received emergency authorization to treat COVID-19 [27]. Similarly, hydroxychloroquine (HCQ) and its precursor compound chloroquine, which was initially used for treating autoimmune diseases and malaria, were repurposed for the treatment of COVID-19, either alone or in combination with azithromycin, with the aim of reducing cytokine production. However, their use was later withdrawn because of concerns about cardiotoxicity [28, 29]. Reports indicate that while de novo drug development typically takes 10–17 years, repurposed drugs can reach the market in 3–12 years, at roughly half the cost [30].
Our review summarizes the latest advances achieved by repurposing drugs that have demonstrated promise in both small-scale studies and clinical trials. We categorized small-molecule inhibitors according to their targeted signaling pathways, focusing on transcription factors, such as NF-κB and AP-1, which are upregulated in chronic conditions. Furthermore, we explored repurposed drugs that are tailored to address specific chronic inflammatory diseases.
Compounds targeting the NF-κB pathway - NF-κB, a pivotal transcription factor, is rapidly activated in various immune cells, encompassing both the innate and adaptive branches, triggered by diverse signals [31]. These signals emanate from innate pattern recognition receptors (PRRs), T-cell receptors (TCRs), B-cell receptors (BCRs), proinflammatory cytokine receptors, and other cellular signaling pathways [32]. The rapid activation of NF-κB in response to these stimuli underscores its central role in orchestrating immune responses across different facets of the immune system [33]. NF-κB functions as either a homodimer or a heterodimer. Five structurally related proteins are critical for NF-κB formation. These genes includes p50/p105, p52/p100, p65 (RelA), c-Rel, and RelB. These dimers are involved in the intricate signal transduction cascade of inflammatory signaling [34]. In the Toll-like receptor 4 (TLR4) signaling pathway, TLR4 recruits toll/interleukin-1 (IL-1) receptor adaptor protein (TIRAP) and TRIF-related adaptor molecule (TRAM or TICAM2) [9].
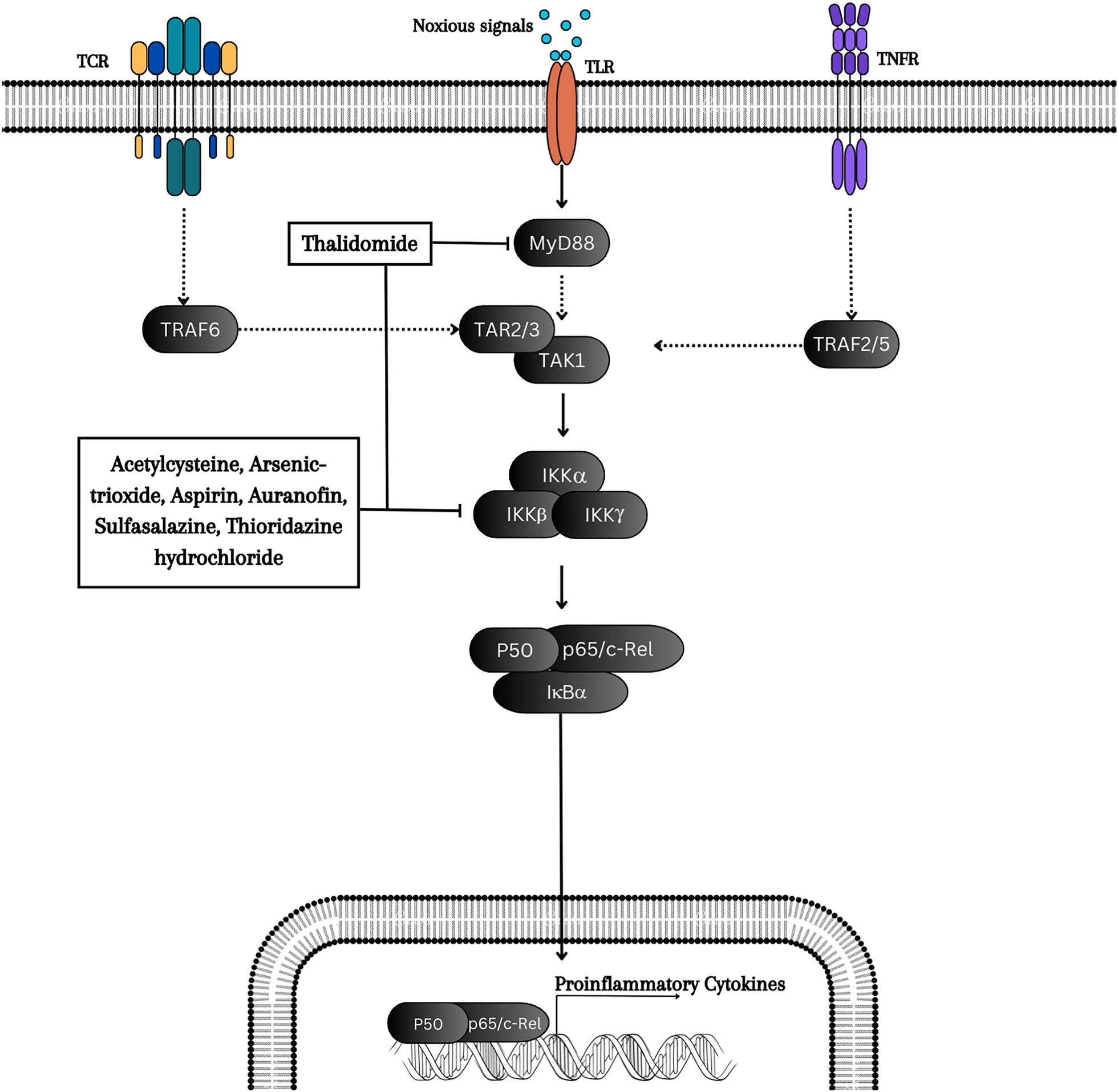
In turn, TIRAP and TRAM recruit myeloid differentiation primary response gene 88 (MyD88) and TIR domain-containing adaptor inducing interferon-beta (TRIF or TICAM1), respectively, for downstream signaling events [35]. Signals from MyD88 lead to the recruitment of IL-1 receptor-associated kinases 1 and 4 (IRAK1 and IRAK4), TNF receptor-associated factor 6 (TRAF6), and TGF-β-activated kinase 1 (TAK1) [36]. TRAF6, which functions as an E3 ubiquitin ligase, mediates autoubiquitination and forms a complex with TAK1 binding protein 2 (TAB2), TAB3, and TAK1. This complex undergoes autophosphorylation and activates TAK1. Subsequently, TAK1 phosphorylates and activates IκB kinases (IKKs), initiating the downstream canonical NF-κB pathway [37]. This cascade ultimately results in the expression of proinflammatory cytokine genes, such as TNF, IL-1, IL-6, IFN, chemokines, and antimicrobial peptides [38]. NF-κB signaling, which serves as a central mediator of inflammation, has been implicated in the broader pathology of numerous diseases, including cancer, rheumatoid arthritis (RA), multiple sclerosis (MS), inflammatory bowel disease (IBD), and atherosclerosis [39,40,41]. Various strategies have been devised to inhibit NF-κB signaling, such as targeting the IKK complex, receptors, or the ubiquitin‒proteasome system to prevent degradation [42]. Below are examples of repurposed small-molecule inhibitors, along with their original target proteins (Table 1), that have demonstrated significant potential in modulating NF-κB activity by targeting specific signaling proteins within the NF-κB pathway, as depicted in Fig. 1.
Fig. 1
Repurposed small-molecule inhibitors of the NF-κB signaling pathway. The canonical pathway begins with activation by Toll-like receptors (TLRs), tumor necrosis factor receptors (TNFRs), and interleukin-1 receptors (IL-1Rs). This activation leads to the phosphorylation and degradation of IκB, allowing NF-κB to translocate into the nucleus and regulate gene expression, thus influencing various cellular processes. Thalidomide inhibits the MyD88 protein, whereas acetylcysteine, arsenic trioxide, aspirin, auranofin, sulfasalazine, and thioridazine hydrochloride act as inhibitors of IKKβ
Table 1 Repurposed small-molecule inhibitors targeting NF-κB signalingAcetylcysteine-N-acetyl-L-cysteine, a derivative of L-cysteine, is a GSH precursor that can regulate the glutathione (GSH)/oxidized (GSSG) glutathione ratio in cells [43]. It has been marketed as an “Acetadote” and was initially approved by the FDA as a mucolytic agent for chronic obstructive pulmonary disease and for addressing acetaminophen (APAP) overdose-induced liver damage by restoring cellular GSH levels [44]. Its anti-inflammatory properties become evident when N-acetyl-L-cysteine decreases TNF-α, IL-1β, IL-6, and IL-10 production in LPS-activated macrophages exposed to mild oxidative stress [45]. N-acetyl-L-cysteine also inhibits the IL-18-stimulated production of TNF-α and IL-6 in vascular smooth muscle cells [46] In LPS-challenged piglet mononuclear phagocytes, N-acetyl-L-cysteine has an anti-inflammatory effect because of its ability to decrease the mRNA expression of NLRP3 and, consequently, IL-1β and IL-18 production [47]. Thus, N-acetyl-L-cysteine exhibits anti-inflammatory effects in in vitro experiments with LPS-stimulated macrophages. However, in in vivo mouse models of LPS inhalation, the drug is less effective or even promotes proinflammatory cytokine production, indicating that either its in vivo availability may be compromised or that there is a complex interplay between several unidentified signaling pathways in the in vivo settings.
Arsenic trioxide-arsenic trioxide has recently been approved by the FDA for the treatment of acute promyelocytic leukemia (APL). However, the compound has a long history dating back over 2400 years to ancient Rome and Greece, where it served as both a poison and therapeutic agent for ailments such as antiseptic, antiperiodic, and antispasmodic ailments [48]. Its anticancer efficacy involves targeting PML–RARα degradation, as demonstrated by Davison et al. [49]. Its anti-inflammatory mechanism involves the inhibition of IKKβ phosphorylation, potentially through interaction with its Cys179 residue, according to Kapahi et al. [50]. Additionally, inhibition of JNK by arsenite has been shown to affect downstream expression, particularly the protein GADD45, a cell cycle inhibitory protein, as demonstrated by Chen et al. [51]. While it has shown promise as an anti-inflammatory agent by inhibiting IκB degradation and subsequent NF-κB activation at a concentration of 12.5 μM and AP-1 activation, its use in humans is a topic of concern because of its dose-dependent toxic effects.
Aspirin, or acetylsalicylic acid, is a notable achievement in the realm of drug discovery as a synthetic compound derived from the parent compound salicylic acid [52]. Originally employed as an analgesic agent, subsequent publications and numerous trials have established its efficacy in preventing myocardial infarction, mitigating migraines, acting as an antiplatelet agent [53], and offering potential benefits in preventing dementia [54, 55]. The mechanism of action of aspirin involves the acetylation of cyclooxygenases in megakaryocytes. This acetylation inhibits the production of platelet thromboxane A2 (TXA2), providing protection against arterial thrombosis, tumor progression, and metastasis [56,57,58]. Additionally, aspirin contributes to the deceleration of atherosclerosis progression by ameliorating endothelial dysfunction and preventing the oxidative modification of low-density lipoprotein (LDL). In colorectal cancer, aspirin acts through diverse mechanisms. It inhibits crucial signaling pathways, such as the NF-kB pathway, by binding to IKK-β, preventing the phosphorylation of downstream proteins [59,60,61,62]. Moreover, aspirin hampers extracellular signal-regulated kinase (ERK) signaling by impeding the binding of c-Raf to Ras [63]. It is also believed to induce apoptosis through mitochondrial pathways and inhibit the Wnt/β-catenin pathway by promoting the phosphorylation and breakdown of β-catenin [64,65,66]. Studies have shown that aspirin and its derivatives inhibit NF-κB and AP-1 activation, particularly at high doses, as evidenced by numerous in vivo models [67]. These multifaceted actions underscore the versatility of aspirin in addressing various health conditions beyond its initial use as an analgesic, thereby highlighting its broad applicability as a therapeutic agent.
Auranofin (AF), initially FDA-approved in 1985 as an oral gold-based antirheumatoid agent, exerts its therapeutic effect by inhibiting thioredoxin reductase (TrxR) and thioredoxin glutathione reductase (TGR) enzymes in rheumatoid arthritis, thus controlling reactive oxygen species (ROS) and DNA damage. It also targets the ubiquitin‒proteasome system in cancer cells [68,69,70,71,72]. It has been repurposed as an anti-inflammatory drug that targets IKK-β, an intermediary in inflammatory cascades, and has been supported by studies conducted by Jeon et al. and Yamashita [73, 74]. It modulates TNF-α, IL-8, and IL-6 secretion by macrophages and monocytes [75]. Computational analysis by Hwangbo et al. suggested that auranofin may disrupt the interaction between lipopolysaccharide (LPS) and Toll-like receptor 4 (TLR4) by targeting the LPS-binding domain, particularly the Arg434 residue. This interference potentially attenuates the proinflammatory response observed in RAW 264.7 macrophages [76]. Furthermore, Hu et al. reported that auranofin acts as an inhibitor of the ubiquitination process involving IκB, functioning as a UCHL5 and USP14 deubiquitinase inhibitor associated with the 19S proteasome [77]. In addition to NF-κB activation, it affects MAPKs, reduces STAT-3, and inhibits angiogenesis [78]. The proinflammatory effects attributed to auranofin are not solely attributed to its interaction with a single intermediate protein such as IKKβ but rather involve multiple proteins, including TLR4 and IκB. This multifactorial mechanism has been corroborated by a combination of in vitro, in vivo, and in silico studies.
Sulfasalazine-Sulfasalazine, an FDA-approved synthetic small-molecule inhibitor used for treating inflammatory bowel disease and rheumatoid arthritis, is derived from the precursor antibiotic and anti-inflammatory agents sulfapyridine and 5-aminosalicylic acid. As noted by Weber et al., the primary target of sulfasalazine is IKKβ in the NF-κB signaling pathway, resulting in a reduction in the levels of proinflammatory cytokines, such as IL-1, IL-6, IL-12, and TNF-α [79,80,81]. Additionally, sulfasalazine inhibits caspase-8 and the expression of receptor activator of nuclear factor kappa-Β ligand (RANKL) [82]. Initially, used for the treatment of ileitis and colitis, it targets folate recognition sites shared by enzymes such as dihydrofolate reductase, serine transhydroxymethylase, and methylenetetrahydrofolate [83]. Kang et al. reported that sulfasalazine inhibited IL-12 production, increased IL-4 production, and decreased IFN-γ production in LPS-stimulated macrophages by binding to the p40-κB site [84]. Thus, sulfasalazine and its analogues exert anti-inflammatory effects by attenuating the activation of the NF-κB pathway [85].
Thalidomide-Thalidomide was initially prescribed as a sedative, tranquilizer, and antiemetic for morning illness [86]. However, its market withdrawal was ensued because of concerns over the adverse effects on fetal development attributed to its binding to the cereblon (CRBN) protein [86,
留言 (0)